Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
AXL is a member of the TAM family of receptor tyrosine kinases (RTKs) that also includes TYRO3 and MER. This was the last family of RTKs to be identified and because their inactivation in mice resulted in rather mild phenotypes, their biological roles were slow to be characterized.
- AXL
1. Introduction
AXL is a member of the TAM family of receptor tyrosine kinases (RTKs) that also includes TYRO3 and MER. This was the last family of RTKs to be identified and because their inactivation in mice resulted in rather mild phenotypes, their biological roles were slow to be characterized. Now, however, in addition to being recognized as key regulators of immune cell activation, TAM RTKs have been shown to be expressed in cancer cells where they promote survival and invasion and contribute to resistance to various therapies. The structure shared by these RTKs includes an extracellular domain consisting of tandem repeats of immunoglobulin-like and fibronectin type 3 (FN-III)-like domains, a single-pass transmembrane domain and an intracellular domain that includes a catalytically competent kinase defined by a unique KWIAIES conserved sequence [1]. While a few genetic mutations and amplifications have been reported in TAMs in cancers (listed in [2]), their functional importance has yet to be defined.
2. AXL Activation and Therapeutic Targeting
AXL can be activated in various ways and these activation mechanisms are unique. Thus, several therapeutic targeting strategies have been designed and evaluated to efficiently inhibit AXL.
2.1. Ligand-Dependent and Ligand-Independent Mechanisms of Activation
The principal ligands that lead to TAM activation are growth arrest specific factor 6 (GAS6) and protein S, both of which require vitamin-K dependent γ-carboxylation to achieve maximal activation [9,10]. These ligands vary in their affinity for the different TAM receptors: GAS6 binds all TAM receptors, with the highest affinity for AXL, while protein S only binds MER and TYRO3. These ligands can also bind the lipid moiety of phosphatidylserine (PS) and can activate TAMs when exposed on apoptotic cells, aggregating platelets, exosomes or virus envelopes [11,12,13]. It has also been shown that GAS6 activation of AXL is localized at regions with high GAS6 concentration, resulting in a diffusional influx of AXL and receptor aggregation and dimerization [14]. Furthermore, GAS6 can mediate dimer formation of other TAM members (MER or TYRO3), which have a lower affinity for GAS6 binding, and more experiments are needed to determine if the receptors can also heterodimerize and how this affects their signaling [15,16,17]. Many laboratory studies have shown that the GAS6/AXL axis promotes cell invasion, proliferation and survival, thus contributing to cancer progression and metastasis. Conversely, clinical studies have shown a correlation between GAS6 expression and patients’ survival in breast cancer, suggesting that GAS6 may not be essential in this context [18,19]. In support of this, it was observed that GAS6 is dispensable in a pre-clinical model of HER2+ breast cancer for the formation of metastases [3]. Interestingly, AXL can be activated in a ligand-independent manner in many pathological contexts. For example, its increased expression, or the presence of oxidative stress, can lead to AXL homodimerization and autophosphorylation [20,21,22]. Furthermore, AXL has been shown to crosstalk and heterodimerize or cluster with other RTKs such as EGFR, HER2, HER3, MET, PDGFR and VEGFR-2 to promote downstream signaling [3,23,24,25,26,27,28,29]. These partnerships serve to diversify the downstream signaling of these RTKs and confer advantages to cancer cells. Through such a mechanism, AXL can promote resistance to a variety of therapies, including chemotherapy and targeted therapies including inhibitors of its partners EGFR, HER2 and PDGF [23,30,31,32,33,34,35,36].
2.2. AXL Therapeutic Targeting
AXL is a particularly interesting candidate as a therapeutic target because its genetic deletion or pharmacological inhibition in mice is well tolerated. Various approaches to inhibit AXL have been studied for cancer treatments including small molecule inhibitors that compete with ATP-binding or monoclonal antibodies [37,38,39]. Indeed, a number of clinical trials are ongoing with the AXL inhibitor R428 (also known as BGB324 or Bemcentinib), which is highly specific for AXL among the TAMs and other RTKs [39]. Additionally, a number of other broad spectrum kinase inhibitors that would also inhibit AXL are currently under study and have been reviewed in [40]. Other approaches have also been envisioned to inhibit the GAS6/AXL axis, including decoy receptors that trap GAS6 and Vitamin K antagonists that would reduce the ability of GAS6 to activate AXL [41,42,43]. However, because there are multiple ways to activate AXL as previously mentioned, there may not be an absolute requirement for GAS6 in certain contexts. More recently, an original approach exploited AXL as a cancer antigen for chimeric antigen receptor (CAR)-T cell therapy. In triple-negative breast cancer (TNBC) where AXL is overexpressed, engineered T cells with AXL-CAR-T were able to induce cytokine release and an antigen-specific cytotoxicity [44,45].
Before we can apply AXL-targeting therapeutic strategies to their full potential in cancer treatment, it is important to better understand the consequences of effective, systemic AXL inhibition. As such, a more detailed understanding of the functions of AXL in various tumor cell types, including both in cancer and different stromal cells, is needed. Accordingly, the next sections will discuss our current understanding of AXL function in a variety of cancer-related biological processes that are both intrinsic and extrinsic to cancer cells.
3. Cancer Cell Intrinsic Implications of AXL Expression in the Metastatic Cascade and Therapy Resistance
Increased AXL expression has been shown to correlate with decreased patients’ survival and metastasis of cancer cells in a plethora of solid cancers. Several biological triggers within the tumors that are known to promote metastasis, such as TGF-β/EMT and hypoxia, impact AXL expression levels in cancer cells [3,4,46,47,48]. Signals downstream of AXL advantage cancer cells throughout the metastatic process and lead to therapy resistance. AXL has been implicated in many steps of the metastatic cascade and recent studies have clarified the cancer cell-intrinsic AXL contributions to this complex process (Figure 1).
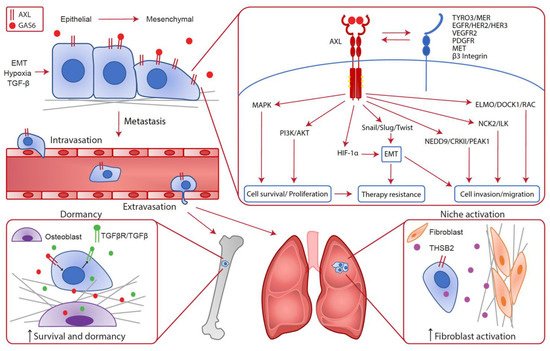
Figure 1. Cancer cell intrinsic implication of AXL in the metastatic cascade. In cancer cells, AXL expression can be enhanced by EMT, hypoxia and TGF-β leading to metastatic progression. Indeed, various cellular pathways downstream of AXL confer advantages to cancer cells such as survival, proliferation, cell invasion and migration, EMT and therapy resistance. Consequently, AXL is required throughout the metastatic cascade for local invasion, intravasation, extravasation and metastatic growth at distant sites. In the bones, AXL expression on disseminated cells increases the survival and dormancy of cancer cells by stimulating the expression of TGF-β2 and TGF-βRs. In the lungs, AXL expression in cancer cells is linked to THSB2 secretion, which activates the fibroblasts in the metastatic niche to promote metastatic growth.
3.1. Epithelial-to-Mesenchymal Transition (EMT)
An emerging function of AXL during cancer progression is its role in the plastic and dynamic program called epithelial-to-mesenchymal transition (EMT). EMT affords several advantages to tumor cells by enhancing cell invasion, apoptosis resistance and stem-like characteristics at the expense of proliferation, therefore contributing to tumor progression and therapy resistance [49]. AXL expression correlates strongly with EMT markers in various cancers [50]. It is now well established that EMT induces the expression of AXL via different EMT transcription factors like SNAIL, SLUG, TWIST or ZEB2 [4,46]. As a feedback mechanism, AXL activation then upregulates the expression of EMT transcription factors and mesenchymal markers to sustain EMT [38,51,52]. Thus, AXL plays multiple roles in tumor progression and metastasis through its modulation of several steps of the metastatic cascade and therapy resistance.
3.2. Cell Invasion and Migration
Invasive characteristics of cancer cells are particularly important in the first steps of the metastatic cascade including invasion of the tumor parenchyma and transendothelial migration during the entry (intravasation) and exit (extravasation) of the systemic circulation. In addition to the role of AXL in enhancing cell invasion by promoting mesenchymal features, other elements triggered by AXL signaling are also important to promote cell motility. For example, AXL can activate several generic downstream effectors from such RTKs as MAPK, PI3K/AKT and JAK/STAT to promote actin reorganization, migration and survival of cancer cells [53,54,55,56,57,58]. Indeed, AXL has been shown to regulate cell adhesion via NCK2/ILK and this signaling converges on the integrin β3 pathway to stimulate cell adhesion [59,60]. In addition, AXL was shown to phosphorylate ELMO proteins in complex with the GEF DOCK1, leading to RAC-mediated cytoskeleton remodeling and migration of TNBC cells [61]. More recently, the first phosphoproteome of AXL was generated following stimulation of AXL by GAS6 in TNBC cells to uncover specific AXL effectors. This phosphoscreen identified NEDD9/CRKII/PEAK1 downstream of AXL and described a novel role for AXL in focal adhesion turnover [62]. This screen also suggested the existence of many other processes and pathways affected by AXL signaling such as RNA transport and vesicle trafficking. Additional research is required to determine which of these AXL-mediated signaling events contributes to invasion and metastasis.
AXL has also been identified as a direct HIF target gene in clear cell renal cell carcinoma (ccRCC) [47]. During hypoxic stress, ccRCC cells upregulate AXL to promote metastasis by maximizing invasion via the GAS6/AXL signaling cascade leading to MET activation. AXL was also shown to regulate HIF-1α levels, thus contributing to the hypoxic response in HER2+ breast cancer cells [48]. In this context, AXL is required for hypoxia-induced EMT and invasion leading to metastasis.
In human cell lines, AXL expression seems to be restricted to invasive and mesenchymal lines, raising the possibility that AXL is a marker for mesenchymal cancers, like TNBC [63]. Nevertheless, its expression in breast cancer correlates with patients’ survival and metastasis independently of the subtype [3]. For example, AXL was found to correlate with metastasis in HER2+ breast cancer, a subtype that retains epithelial characteristics even though it often leads to metastasis and evidence suggests that EMT is required for this capacity [3,64]. Indeed, during progression of epithelial cancer, only a small population of motile cells invade the surrounding tissue to disseminate and form metastases. TGF-β, a factor that induces EMT, is required in several steps of the metastatic process including local invasion, intravasation and extravasation [65,66,67]. Interestingly, in epithelial cell lines that do not express AXL at basal levels, like HER2+ cells and patient-derived xenografts (PDX), TGF-β reprogramming can promote AXL expression [3,68]. In this context, AXL deletion in a preclinical model of HER2+ breast cancer almost completely abrogated the metastatic dissemination by blocking intravasation, extravasation and growth at the metastatic site [3], supporting a role for AXL in TGF-β-induced cell invasion.
These new findings regarding AXL signaling, and biology further confirm its role in cancer cell dissemination through the reprogramming of cells to be able to leave the primary tumor and enter a distant organ by increasing cell motility.
3.3. Modulation of the Metastatic Niche and Dormancy
Following entry in a distant organ, cells must adapt to the foreign environment to form macrometastases. To do so, mesenchymal cells must be reprogrammed to a more epithelial state to be able to proliferate. In a HER2+ breast cancer model, inducible AXL downregulation after the arrival of cancer cells in the lungs led to a reduction of metastatic outgrowth [3]. A similar observation was made using cells isolated from the PyMT murine breast cancer model and with MDA-MB-231 breast cancer cells where it was found that AXL inhibition post-seeding reduced the metastatic burden [69]. By using AXL+ tumor initiating cells that display partial EMT, it was suggested that AXL prepared the metastatic niche by activating lung fibroblasts via the secretion of THSB2. Those fibroblasts subsequently promoted a switch toward a more epithelial state, reducing AXL levels and promoting proliferation of cancer cells leading to the formation of macrometastasis.
Disseminated cancer cells can also enter a dormant state in the new organ instead of proliferating and can then survive in a quiescent state for years where they are highly resistant to treatment and can lead to relapses in patients [70]. Both AXL and GAS6 have been implicated in dormancy in the context of bone marrow metastasis of prostate cancer. In the bone marrow niche, osteoblasts produce GAS6 that can activate AXL on the surface of nearby disseminated cancer cells [71,72]. AXL activation then induces the expression of TGF-β2 and TGF-βRs that results in the arrest of proliferation and cell survival [72]. AXL has also been reported to be strongly expressed in dormant myeloma cells and its inhibition induces their proliferation [73]. Altogether, these results suggest that the persistence of AXL expression and signaling in disseminated cancer cells can lead to dormancy in the metastatic niche in some cancers.
3.4. Therapy Resistance
AXL promotes and sustains a mesenchymal phenotype leading to metastasis, but its role in EMT also has an important impact on therapy outcome. Indeed, EMT is associated with a wide range of changes linked to stemness and drug resistance, implicating AXL as an important mediator of resistance to chemotherapy, antimitotic drugs, and various targeted therapies.
In support of this role, AXL has been found to be upregulated in chemo-resistant cells in a variety of cancers. Many studies have shown a better drug sensitivity when combining AXL inhibition with chemotherapeutic compounds such as docetaxel, cisplatin, pemetrexel, vincristine, paclitaxel, adryamicin, gemcitabine or carboplatin [74,75,76,77,78]. In cancer cells, chemotherapy induces EMT and AXL is also upregulated in this context [51,56,68]. Indeed, AXL was found to be associated with mesenchymal features of breast and lung cancer cells and its inhibition synergized with antimitotic agents to induce cell death [68]. Furthermore, genetic, or pharmacologic inhibition of AXL was shown to revert EMT in pancreatic and prostate cancer cells and to modulate the expression of nucleoside transporters playing a role in chemotherapeutic response [75,77]. Additionally, many studies link resistance to the EGFR inhibitor Erlotinib to EMT and AXL overexpression [57,79]. In this context, erlotinib-resistant cells displayed EMT features that were prevented by AXL inhibition. However, EMT-associated resistance to erlotinib has also been reported to be independent of AXL [68] and further studies are needed to explain this discrepancy. Crizotinib, an ALK inhibitor, has also been linked to the activation of AXL and induction of EMT [80]. Thus, interfering with AXL can improve responses to various therapies by reverting EMT and dampening downstream pathways that lead to drug resistance. In conclusion, numerous studies provide rationale for the use of AXL inhibition to enhance sensitivity and/or prevent resistance to cytotoxic chemotherapies and targeted agents, highlighting the potential of AXL-targeted drugs in combination with a variety of other therapies.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14030466
This entry is offline, you can click here to edit this entry!