Nitric oxide (NO) is a highly reactive gas with a brief life span, synthesized by the enzyme nitric oxide synthase (NOS) through L-arginine oxidation to L-citrulline. The dual role of NO in embryonic stem cells (ESCs) has been previously reported, preserving pluripotency and cell survival or inducing differentiation with a dose-dependent pattern. In this line, high doses of NO have been used in vitro cultures to induce focused differentiation toward different cell lineages being a key molecule in the regenerative medicine field. Moreover, optimal conditions to promote pluripotency in vitro are essential for their use in advanced therapies.
- nitric oxide
- stem cell
- evolution
- cell signaling
- cell differentiation
1. Biological Functions of Nitric Oxide
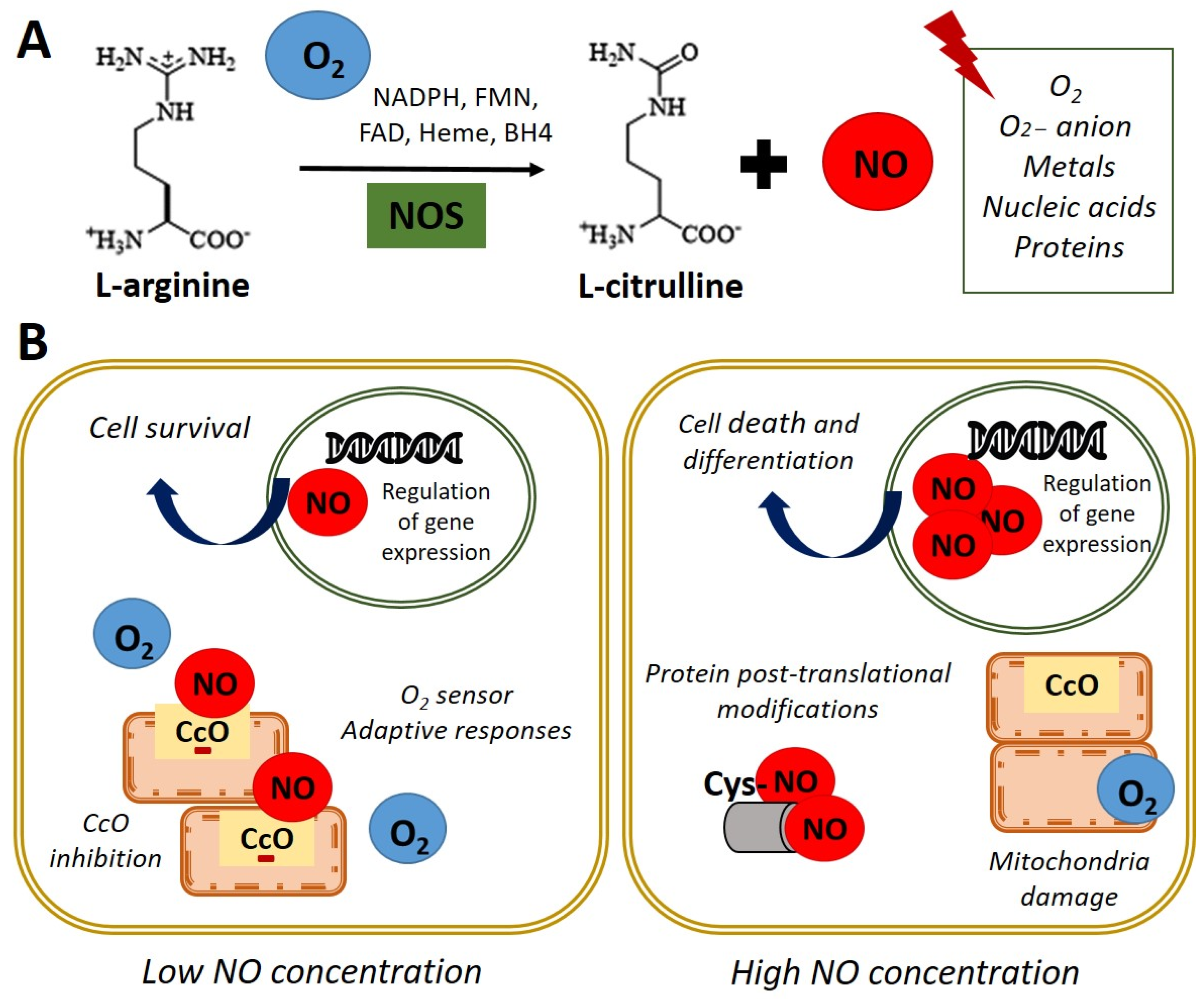 Figure 1. Nitric oxide synthesis and biological functions. (A) In normoxia conditions (21% O2), nitric oxide synthase (NOS) catalyzes the oxidation of the terminal guanidinyl nitrogen of the amino acid L-arginine to form L-citrulline and nitric oxide (NO) in presence of NADPH and cofactors such as flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), heme, and tetrahydrobiopterin (BH4) [3]. Once produced, NO readily interacts with O2, O2− anion, metals, nucleic acids, and proteins. (B) Left panel. NO at low concentration inhibits cytochrome c oxidase (CcO) activity by competing with O2. Adaptive responses to O2 concentration and cell survival genes are activated. Right panel. High concentrations of NO induce damage in all mitochondrial complexes, nitrosylation, or oxidation of protein thiol groups and induce cell death and differentiation.
Figure 1. Nitric oxide synthesis and biological functions. (A) In normoxia conditions (21% O2), nitric oxide synthase (NOS) catalyzes the oxidation of the terminal guanidinyl nitrogen of the amino acid L-arginine to form L-citrulline and nitric oxide (NO) in presence of NADPH and cofactors such as flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), heme, and tetrahydrobiopterin (BH4) [3]. Once produced, NO readily interacts with O2, O2− anion, metals, nucleic acids, and proteins. (B) Left panel. NO at low concentration inhibits cytochrome c oxidase (CcO) activity by competing with O2. Adaptive responses to O2 concentration and cell survival genes are activated. Right panel. High concentrations of NO induce damage in all mitochondrial complexes, nitrosylation, or oxidation of protein thiol groups and induce cell death and differentiation.2. Nitric Oxide Signaling Pathways in Stem Cell
3. Nitric Oxide in the Embryonic Development
4. Nitric Oxide and Stemness of Pluripotent Cells
4.1. Nitric Oxide and Stem Cell Differentiation
4.2. Nitric Oxide and Pluripotency
This entry is adapted from the peer-reviewed paper 10.3390/antiox11030497
References
- Marletta, M.A. Nitric oxide synthase: Aspects concerning structure and catalysis. Cell 1994, 78, 927–930.
- Cosby, K.; Partovi, K.S.; Crawford, J.H.; Patel, R.P.; Reiter, C.D.; Martyr, S.; Yang, B.K.; Waclawiw, M.A.; Zalos, G.; Xu, X.; et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 2003, 9, 1498–1505.
- Bredt, D.S. Endogenous nitric oxide synthesis: Biological functions and pathophysiology. Free Radic. Res. 1999, 31, 577–596.
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142.
- Tejedo, J.R.; Tapia-Limonchi, R.; Mora-Castilla, S.; Cahuana, G.M.; Hmadcha, A.; Martin, F.; Bedoya, F.J.; Soria, B. Low concentrations of nitric oxide delay the differentiation of embryonic stem cells and promote their survival. Cell Death Dis. 2010, 1, e80.
- Mora-Castilla, S.; Tejedo, J.R.; Hmadcha, A.; Cahuana, G.M.; Martín, F.; Soria, B.; Bedoya, F.J. Nitric oxide repression of Nanog promotes mouse embryonic stem cell differentiation. Cell Death Differ. 2010, 17, 1025–1033.
- Krumenacker, J.S.; Hanafy, K.A.; Murad, F. Regulation of nitric oxide and soluble guanylyl cyclase. Brain Res. Bull. 2004, 62, 505–515.
- Tanioka, T.; Tamura, Y.; Fukaya, M.; Shinozaki, S.; Mao, J.; Kim, M.; Shimizu, N.; Kitamura, T.; Kaneki, M. Inducible nitric-oxide synthase and nitric oxide donor decrease insulin receptor substrate-2 protein expression by promoting proteasome-dependent degradation in pancreatic beta-cells: Involvement of glycogen synthase kinase-3beta. J. Biol. Chem. 2011, 286, 29388–29396.
- Bloch, W.; Fleischmann, B.K.; Lorke, D.E.; Andressen, C.; Hops, B.; Hescheler, J.; Addicks, K. Nitric oxide synthase expression and role during cardiomyogenesis. Cardiovasc. Res. 1999, 43, 675–684.
- Brown, G.C.; Cooper, C.E. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994, 356, 295–298.
- Foster, M.W.; Stamler, J.S. New insights into protein S-nitrosylation. Mitochondria as a model system. J. Biol. Chem. 2004, 279, 25891–25897.
- Mateo, J.; García-Lecea, M.; Cadenas, S.; Hernández, C.; Moncada, S. Regulation of hypoxia-inducible factor-1α by nitric oxide through mitochondria-dependent and -independent pathways. Biochem. J. 2003, 376, 537–544.
- Almeida, A.; Moncada, S.; Bolaños, J.P. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat. Cell Biol. 2004, 6, 45–51.
- Caballano-Infantes, E.; Díaz, I.; Hitos, A.B.; Cahuana, G.M.; Martínez-Ruiz, A.; Soria-Juan, B.; Rodríguez-Griñolo, R.; Hmadcha, A.; Martín, F.; Soria, B.; et al. Stemness of human pluripotent cells: Hypoxia-like response induced by low nitric oxide. Antioxidants 2021, 10, 1408.
- Dawson, T.M.; Dawson, V.L. Nitric Oxide Signaling in Neurodegeneration and Cell Death. Adv. Pharmacol. 2018, 82, 57–83.
- Cahuana, G.M.; Tejedo, J.R.; Hmadcha, A.; Ramírez, R.; Cuesta, A.L.; Soria, B.; Martin, F.; Bedoya, F.J. Nitric oxide mediates the survival action of IGF-1 and insulin in pancreatic β cells. Cell. Signal. 2008, 20, 301–310.
- Lundberg, J.O.; Gladwin, M.T.; Ahluwalia, A.; Benjamin, N.; Bryan, N.S.; Butler, A.; Cabrales, P.; Fago, A.; Feelisch, M.; Ford, P.C.; et al. Nitrate and nitrite in biology, nutrition and therapeutics. Nat. Chem. Biol. 2009, 5, 865–869.
- Salguero-Aranda, C.; Tapia-Limonchi, R.; Cahuana, G.M.; Hitos, A.B.; Diaz, I.; Hmadcha, A.; Fraga, M.; Martín, F.; Soria, B.; Tejedo, J.R.; et al. Differentiation of Mouse Embryonic Stem Cells toward Functional Pancreatic β-Cell Surrogates through Epigenetic Regulation of Pdx1 by Nitric Oxide. Cell Transplant. 2016, 25, 1879–1892.
- Krumenacker, J.S.; Katsuki, S.; Kots, A.; Murad, F. Differential expression of genes involved in cGMP-dependent nitric oxide signaling in murine embryonic stem (ES) cells and ES cell-derived cardiomyocytes. Nitric Oxide Biol. Chem. 2006, 14, 1–11.
- Krumenacker, J.S.; Murad, F. NO-cGMP signaling in development and stem cells. Mol. Genet. Metab. 2006, 87, 311–314.
- Mujoo, K.; Krumenacker, J.S.; Murad, F. Nitric oxide-cyclic GMP signaling in stem cell differentiation. Free Radic. Biol. Med. 2011, 51, 2150–2157.
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and the ugly. Am. J. Physiol. Cell Physiol. 1996, 271, 1424–1437.
- Murad, F. Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N. Engl. J. Med. 2006, 355, 2003–2011.
- Tranguch, S.; Huet-Hudson, Y. Decreased viability of nitric oxide synthase double knockout mice. Mol. Reprod. Dev. 2003, 65, 175–179.
- Lee, T.C.; Zhao, Y.D.; Courtman, D.W.; Stewart, D.J. Abnormal Aortic Valve Development in Mice Lacking Endothelial Nitric Oxide Synthase. Circulation 2000, 101, 2345–2348.
- Feng, Q.; Song, W.; Lu, X.; Hamilton, J.A.; Lei, M.; Peng, T.; Yee, S.P. Development of heart failure and congenital septal defects in mice lacking endothelial nitric oxide synthase. Circulation 2002, 106, 873–879.
- Cooke, R.M.; Mistry, R.; John Challiss, R.A.; Straub, V.A. Nitric oxide synthesis and cGMP production is important for neurite growth and synapse remodeling after axotomy. J. Neurosci. 2013, 33, 5626–5637.
- Jisha, J.; Schwaerzer, G.K.; Kalyanaraman, H.; Cory, E.; Sah, R.L.; Li, M.; Vaida, F.; Boss, G.R.; Pilz, R.B. Soluble guanylate cyclase as a novel treatment target for osteoporosis. Endocrinology 2014, 155, 4720–4730.
- Kalyanaraman, H.; Schall, N.; Pilz, R.B. Nitric oxide and cyclic GMP functions in bone. Nitric Oxide 2018, 76, 62–70.
- Tesfamariam, B. Targeting heme-oxidized soluble guanylate cyclase to promote osteoblast function. Drug Discov. Today 2020, 25, 422–429.
- Lee, J.S.; Lee, H.J.; Lee, J.W.; Lee, S.C.; Heo, J.S. Osteogenic Effect of Inducible Nitric Oxide Synthase (iNOS)-Loaded Mineralized Nanoparticles on Embryonic Stem Cells. Cell. Physiol. Biochem. 2018, 51, 746–762.
- Mitrovic, V.; Jovanovic, A.; Lehinant, S. Soluble guanylate cyclase modulators in heart failure. Curr. Heart Fail. Rep. 2011, 8, 38–44.
- Breitenstein, S.; Roessig, L.; Sandner, P.; Lewis, K.S. Novel sGC Stimulators and sGC Activators for the Treatment of Heart Failure. Handb. Exp. Pharmacol. 2017, 243, 225–247.
- Armstrong, P.W.; Roessig, L.; Patel, M.J.; Anstrom, K.J.; Butler, J.; Voors, A.A.; Lam, C.S.P.; Ponikowski, P.; Temple, T.; Pieske, B.; et al. A Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial of the Efficacy and Safety of the Oral Soluble Guanylate Cyclase Stimulator: The VICTORIA Trial. JACC Heart Fail. 2018, 6, 96–104.
- Moghaddam, N.; Malhi, N.; Toma, M. Impact of oral soluble guanylate cyclase stimulators in heart failure: A systematic review and Meta-analysis of randomized controlled trials. Am. Heart J. 2021, 241, 74–82.
- Okubo, T.; Fujimoto, S.; Hayashi, D.; Suzuki, T.; Sakaue, M.; Miyazaki, Y.; Tanaka, K.; Usami, M.; Takizawa, T. Valproic acid promotes mature neuronal differentiation of adipose tissue-derived stem cells through iNOS–NO–sGC signaling pathway. Nitric Oxide 2019, 93, 1–5.
- Bandara, N.; Gurusinghe, S.; Kong, A.; Mitchell, G.; Wang, L.X.; Lim, S.Y.; Strappe, P. Generation of a nitric oxide signaling pathway in mesenchymal stem cells promotes endothelial lineage commitment. J. Cell. Physiol. 2019, 234, 20392–20407.
- Reynaert, N.L.; Ckless, K.; Korn, S.H.; Vos, N.; Guala, A.S.; Wouters, E.F.M.; Van Der Vliet, A.; Janssen-Heininger, Y.M.W. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc. Natl. Acad. Sci. USA 2004, 101, 8945–8950.
- Chu, L.; Jiang, Y.; Hao, H.; Xia, Y.; Xu, J.; Liu, Z.; Verfaillie, C.M.; Zweier, J.L.; Liu, Z. Nitric oxide enhances Oct-4 expression in bone marrow stem cells and promotes endothelial differentiation. Eur. J. Pharmacol. 2008, 591, 59–65.
- Chambers, I.; Colby, D.; Robertson, M.; Nichols, J.; Lee, S.; Tweedie, S.; Smith, A. Functional Expression Cloning of Nanog, a Pluripotency Sustaining Factor in Embryonic Stem Cells. Cell 2003, 113, 643–655.
- Chanda, P.K.; Meng, S.; Lee, J.; Leung, H.E.; Chen, K.; Cooke, J.P. Nuclear S-nitrosylation defines an optimal zone for inducing pluripotency. Circulation 2019, 140, 1081–1099.
- Peunova, N.; Scheinker, V.; Cline, H.; Enikolopov, G. Nitric oxide is an essential negative regulator of cell proliferation in Xenopus brain. J. Neurosci. 2001, 21, 8809–8818.
- Enikolopov, G.; Banerji, J.; Kuzin, B. Nitric oxide and Drosophila development. Cell Death Differ. 1999, 6, 956–963.
- Tranguch, S.; Steuerwald, N.; Huet-Hudson, Y.M. Nitric oxide synthase production and nitric oxide regulation of preimplantation embryo development. Biol. Reprod. 2003, 68, 1538–1544.
- Leri, A.; Kajstura, J.; Anversa, P. Role of cardiac stem cells in cardiac pathophysiology: A paradigm shift in human myocardial biology. Circ. Res. 2011, 109, 941–961.
- Tsutsui, M.; Shimokawa, H.; Morishita, T.; Nakashima, Y.; Yanagihara, N. Development of genetically engineered mice lacking all three nitric oxide synthases. J. Pharmacol. Sci. 2006, 102, 147–154.
- Van Voorhis, B.J.; Dunn, M.S.; Snyder, G.D.; Weiner, C.P. Nitric oxide: An autocrine regulator of human granulosa-luteal cell steroidogenesis. Endocrinology 1994, 135, 1799–1806.
- Kagabu, S.; Kodama, H.; Fukuda, J.; Karube, A.; Murata, M.; Tanaka, T. Inhibitory effects of nitric oxide on the expression and activity of aromatase in human granulosa cells. Mol. Hum. Reprod. 1999, 5, 396–401.
- Dineva, J.D.; Vangelov, I.M.; Nikolov, G.G.; Konakchieva, R.T.; Ivanova, M.D. Nitric oxide stimulates the production of atrial natriuretic peptide and progesteron by human granulosa luteinized cells with an antiapoptotic effect. Endocr. Regul. 2008, 42, 45–51.
- Bergandi, L.; Basso, G.; Evangelista, F.; Canosa, S.; Dalmasso, P.; Aldieri, E.; Revelli, A.; Benedetto, C.; Ghigo, D. Inducible nitric oxide synthase and heme oxygenase 1 are expressed in human cumulus cells and may be used as biomarkers of oocyte competence. Reprod. Sci. 2014, 21, 1370–1377.
- Krause, B.J.; Hanson, M.A.; Casanello, P. Role of nitric oxide in placental vascular development and function. Placenta 2011, 32, 797–805.
- Zhang, X.; Shi, J.; Chen, S.; Dong, Y.; Zhang, L.; Midgley, A.C.; Kong, D.; Wang, S. Polycaprolactone/gelatin degradable vascular grafts simulating endothelium functions modified by nitric oxide generation. Regen. Med. 2019, 14, 1089–1105.
- Zhang, X.G.; Jin, L.; Tian, Z.; Wang, J.; Yang, Y.; Liu, J.F.; Chen, Y.; Hu, C.H.; Chen, T.Y.; Zhao, Y.R.; et al. Nitric oxide inhibits autophagy and promotes apoptosis in hepatocellular carcinoma. Cancer Sci. 2019, 110, 1054–1063.
- Cahuana, G.M.; Tejedo, J.R.; Jiménez, J.; Ramírez, R.; Sobrino, F.; Bedoya, F.J. Nitric oxide-induced carbonylation of Bcl-2, GAPDH and ANT precedes apoptotic events in insulin-secreting RINm5F cells. Exp. Cell Res. 2004, 293, 22–30.
- Li, J.; Bombeck, C.A.; Yang, S.; Kim, Y.M.; Billiar, T.R. Nitric oxide suppresses apoptosis via interrupting caspase activation and mitochondrial dysfunction in cultured hepatocytes. J. Biol. Chem. 1999, 274, 17325–17333.
- Kanno, S.; Kim, P.K.M.; Sallam, K.; Lei, J.; Billiar, T.R.; Shears, L.L. Nitric oxide facilitates cardiomyogenesis in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 2004, 101, 12277–12281.
- Krischel, V.; Bruch-Gerharz, D.; Suschek, C.; Kröncke, K.D.; Ruzicka, T.; Kolb-Bachofen, V. Biphasic effect of exogenous nitric oxide on proliferation and differentiation in skin derived keratinocytes but not fibroblasts. J. Investig. Dermatol. 1998, 111, 286–291.
- Størling, J.; Binzer, J.; Andersson, A.K.; Züllig, R.A.; Tonnesen, M.; Lehmann, R.; Spinas, G.A.; Sandler, S.; Billestrup, N.; Mandrup-Poulsen, T. Nitric oxide contributes to cytokine-induced apoptosis in pancreatic beta cells via potentiation of JNK activity and inhibition of Akt. Diabetologia 2005, 48, 2039–2050.
- Tejedo, J.; Bernabé, J.C.; Ramírez, R.; Sobrino, F.; Bedoya, F.J. NO induces a cGMP-independent release of cytochrome c from mitochondria which precedes caspase 3 activation in insulin producing RINm5F cells. FEBS Lett. 1999, 459, 238–243.
- Vodovotz, Y.; Kim, P.K.M.; Bagci, E.Z.; Ermentrout, G.B.; Chow, C.C.; Bahar, I.; Billiar, T.R. Inflammatory Modulation of Hepatocyte Apoptosis by Nitric Oxide: In Vivo, In Vitro, and In Silico Studies. Curr. Mol. Med. 2005, 4, 753–762.
- Tejedo, J.R.; Cahuana, G.M.; Ramírez, R.; Esbert, M.; Jiménez, J.; Sobrino, F.; Bedoya, F.J. nitric oxide triggers the phosphatidylinositol 3-kinase/Akt survival pathway in insulin-producing RINm5F cells by arousing Src to activate insulin receptor substrate-1. Endocrinology 2004, 145, 2319–2327.
- Nie, Y.; Zhang, K.; Zhang, S.; Wang, D.; Han, Z.; Che, Y.; Kong, D.; Zhao, Q.; Han, Z.; He, Z.X.; et al. Nitric oxide releasing hydrogel promotes endothelial differentiation of mouse embryonic stem cells. Acta Biomater. 2017, 63, 190–199.
- Tapia-Limonchi, R.; Cahuana, G.M.; Caballano-Infantes, E.; Salguero-Aranda, C.; Beltran-Povea, A.; Hitos, A.B.; Hmadcha, A.; Martin, F.; Soria, B.; Bedoya, F.J.; et al. Nitric Oxide Prevents Mouse Embryonic Stem Cell Differentiation through Regulation of Gene Expression, Cell Signaling and Control of Cell Proliferation. J. Cell Biochem. 2016, 117, 2078–2088.
- Chung, H.T.; Pae, H.O.; Choi, B.M.; Billiar, T.R.; Kim, Y.M. Nitric Oxide as a Bioregulator of Apoptosis. Biochem. Biophys. Res. Commun. 2001, 282, 1075–1079.
- Yamane, T.; Dylla, S.J.; Muijtjens, M.; Weissman, I.L. Enforced Bcl-2 expression overrides serum and feeder cell requirements for mouse embryonic stem cell self-renewal. Proc. Natl. Acad. Sci. USA 2005, 102, 3312–3317.
- Zhu, Z.; Pan, Q.; Zhao, W.; Wu, X.; Yu, S.; Shen, Q.; Zhang, J.; Yue, W.; Peng, S.; Li, N.; et al. BCL2 enhances survival of porcine pluripotent stem cells through promoting FGFR2. Cell Prolif. 2021, 54, e12932.
- Kim, Y.M.; De Vera, M.E.; Watkins, S.C.; Billiar, T.R. Nitric oxide protects cultured rat hepatocytes from tumor necrosis factor-α-induced apoptosis by inducing heat shock protein 70 expression. J. Biol. Chem. 1997, 272, 1402–1411.
- Mannick, J.B.; Miao, X.Q.; Stamler, J.S. Nitric oxide inhibits Fas-induced apoptosis. J. Biol. Chem. 1997, 272, 24125–24128.
- Wong, J.C.; Fiscus, R.R. Essential roles of the nitric oxide (no)/cGMP/protein kinase G type-Iα (PKG-Iα) signaling pathway and the atrial natriuretic peptide (ANP)/cGMP/PKG-Iα autocrine loop in promoting proliferation and cell survival of OP9 bone marrow stromal cells. J. Cell. Biochem. 2011, 112, 829–839.