1. Initiation of T Cell Signaling
The initial step in T cell activation is the conversion of an extracellular signal by binding of an antigen to its specific TCR, which induces an intracellular signaling cascade (
Figure 1). This is required to switch the T cell from a resting state to proliferation and differentiation and is characterized by intracellular structural changes such as actin reorganization and metabolic reprogramming
[1][2][3]. Structurally, TCR signals are transmitted via the noncovalently associated CD3ζ subunits. These subunits possess intracellular tails containing specific signaling motifs, so-called immunoreceptor tyrosine-based activation motifs (ITAMs). TCR activation results in the phosphorylation of these ITAMs by the two members of the Src-family of protein tyrosine kinases: lymphocyte-specific protein tyrosine kinase Lck and proto-oncogene tyrosine-protein kinase Fyn. Phosphorylated ITAMs provide docking sites for the tandem SH2 domains of the Syk family kinase ζ-chain-associated protein kinase 70 (ZAP70), which is thereby recruited to the plasma membrane and is activated itself via phosphorylation by Lck
[4]. Phosphorylated ZAP70 is the key component for further downstream signaling, which leads to phosphorylation of the linker of activation of T cells (LAT) and Src homoly 2 domain-containing leukocyte protein of 76kDa (SLP-76).
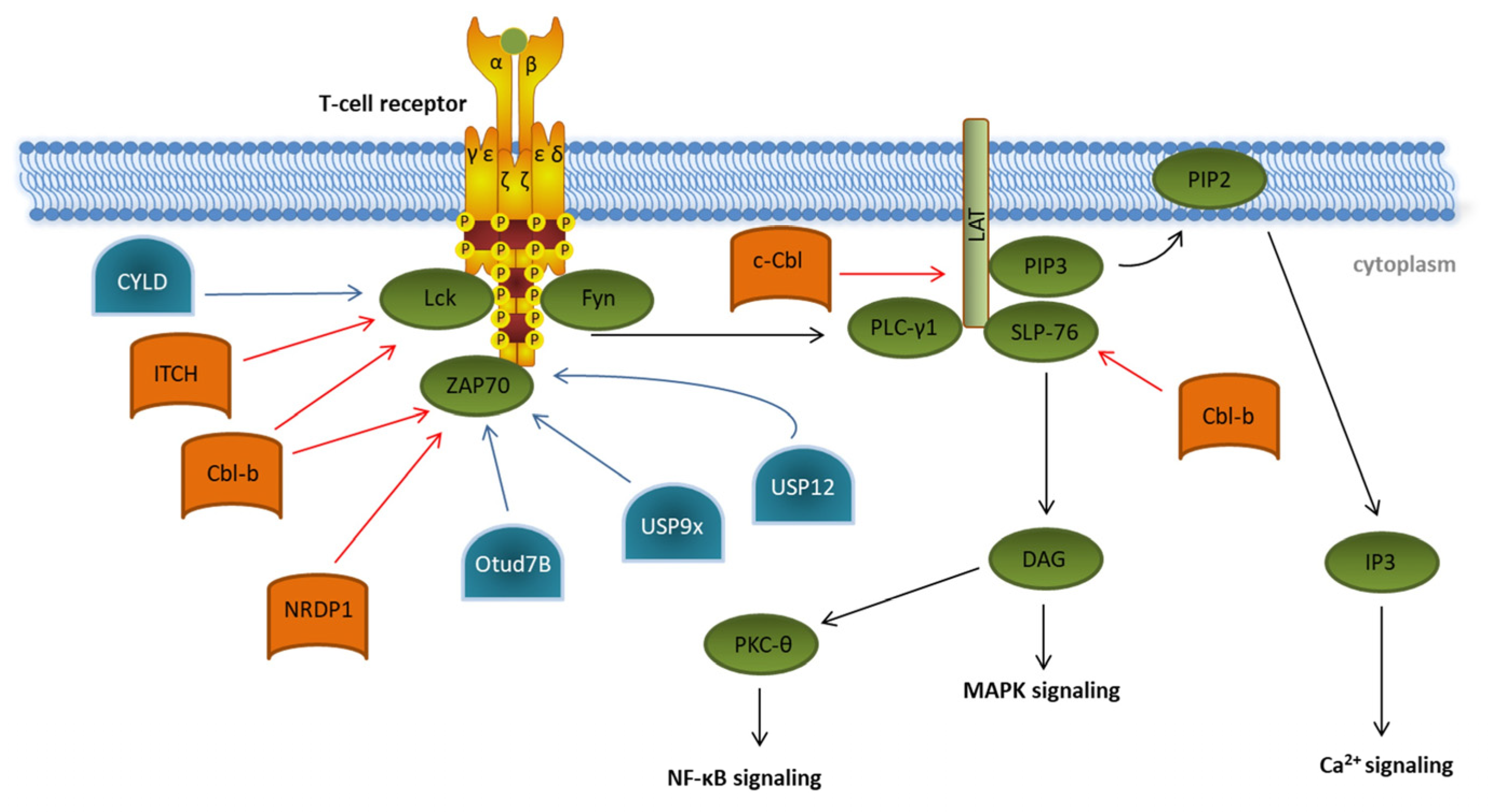
Figure 1. Ubiquitination and deubiquitination in proximal T cell signaling. Overview of known ubiquitin E3 ligases (orange) and deubiquitinating enzymes (blue) involved in regulating the initiation of T cell receptor signaling (green). Details are described in the main text of the manuscript. The T cell signaling pathway is indicated by black arrows. Modifications that dampen T cell receptor signaling are indicated by red arrows. Ubiquitin modifications enhancing T cell signaling are indicated by blue arrows.
In the last decades, several E3 ubiquitin ligases participating in the initiation of T cell signaling have been identified, e.g., the two E3 ligases of the Casitas B-cell Lymphoma (Cbl) protein family, c-Cbl and Cbl-b, and the HECT-type E3 ubiquitin ligase Itchy (ITCH)
[5][6][7]. Cbl-b interacts, via its multiple protein-interacting domains, with key TCR signalosome proteins such as Lck, SLP-76, and ZAP70 and targets them for proteasomal degradation (
Figure 1)
[8][9]. In a cooperative manner with the E3 ligase ITCH, Cbl-b can mediate the K33-linked polyubiquitination of the TCR-ζ chain, which results in reduced ZAP-70 interaction and subsequently decreases downstream phosphorylation
[10]. ZAP70 is also targeted by the neuregulin receptor degradation protein-1 (NRDP1)/RNF41 E3 ligase attaching K33-linked ubiquitin chains to ZAP70 (
Figure 1)
[11]. Both Cbl-b- and NRDP1-dependent ubiquitination of ZAP70 lead to the recruitment of suppressors of T cell signaling 1 and 2 (STS1/2), which dephosphorylate ZAP70, ultimately resulting in an abrogation of downstream signaling. The role of the E3 ligases Cbl-b, ITCH, and NRDP1 in the regulation of T cell activation is further emphasized by data obtained from experiments in KO mice, which showed the hyperproliferation of T cells, reflecting a strong autoimmune phenotype
[10][11][12]. Finally, c-Cbl targets the ZAP70 substrate LAT for ubiquitin-mediated degradation, preventing the formation of the LAT complex and therefore abrogating further phosphorylation events downstream of LAT
[13].
There are a few deubiquitinating enzymes (DUBs) antagonizing these processes. CYLD, a tumor suppressor in familial cylindromatosis, an autosomal-dominant genetic predisposition to multiple tumors of the skin appendages, interacts with active Lck and promotes the recruitment of active Lck to its substrate Zap70
[14]. Other DUB candidates like the ubiquitin-specific peptidases Usp12 and Usp9x are capable of stabilizing the T cell receptor complex at the plasma membrane, leading to prolonged TCR signaling
[15][16]. This was supported by experiments in Usp9x knockout mice pointing to reduced TCR signaling during thymic development, which results in decreased negative selection, ultimately leading to increased numbers of autoreactive T cells
[15].
Furthermore, OTUD7B, which is a member of the ovarian tumor (OTU) family of DUBs, deubiquitinates ZAP70, preventing its association with the suppressors of T cell signaling STS1 and STS2, therefore enabling enhanced T cell activation (
Figure 1)
[17]. This is underlined by the fact that OTUD7B knockout mice are refractory to T cell-mediated autoimmune or inflammatory responses
[17].
Recently, some signaling proteins that are sumoylated during T cell activation have been identified. The signaling protein phospholipase C-γ1 (PLC-γ1), which is phosphorylated by ZAP70 upon T cell receptor activation, is sumoylated by the Sumo-E3 ligase PIASxβ, which appears to be required for its membrane localization to the LAT complex
[18]. In contrast, the desumoylation of PLC-γ1 prevents complex formation and, therefore, blocks further downstream signaling
[18]. Moreover, SLP-76, which is also part of the LAT complex, becomes sumoylated rapidly upon T cell stimulation, and this is crucial for activation of the transcription factor nuclear factor of activated T cells (NFAT)
[19].
Engagement of the TCR and subsequent downstream signaling is accompanied by forming of an immunological synapse at the APC-TCR contact site, resulting in structural and morphological changes bringing surface molecules and intracellular signaling proteins into close proximity. This is important for the successful activation of the T cell signaling pathway
[20]. In this regard, TCR-induced sumoylation of the protein kinase C theta (PKC-θ) is essential for the formation of mature immunological synapses and for T cell activation. The desumoylation of PKC-θ blocks its localization to the immune synapse, resulting in dysregulated activation and the proliferation of T cells
[21].
Although Cbl-b has been shown to be able to neddylate proteins associated with transforming growth factor-β (TGF-β) signaling, no neddylation has yet been identified in proximal T cell signaling. This is underlined by data showing that application of the neddylation inhibitor MLN4924/Pevonedistad has no effect on the phosphorylation of signaling proteins upon T cell activation
[22].
In summary, the initiation of TCR signaling is highly regulated by ubiquitination targeting the TCR-ZAP70 interaction as the main checkpoint. In addition, only a few signaling proteins have been shown to be sumoylated. With respect to the current literature, ubiquitin-like modifiers such as NEDD8, ISG15, or FAT10 are not involved in the proximal T cell signaling cascade.
2. Transcription Factor Activation in T Cells
The induction of T cell-specific cytokine production, cell proliferation, and differentiation requires the translocation of specific transcription factors into the nucleus.
After triggering the T cell receptor, several signaling cascades downstream of ZAP70 are activated; mainly MAPK pathways via ERK and JNK, modulation of the Ca2+ flux, and activation of PKC-θ lead to the activation of the transcription factors NFAT, the nuclear factor “kappa-light-chain-enhancer” of activated B cells, NF-κB, and JunB/AP-1 (
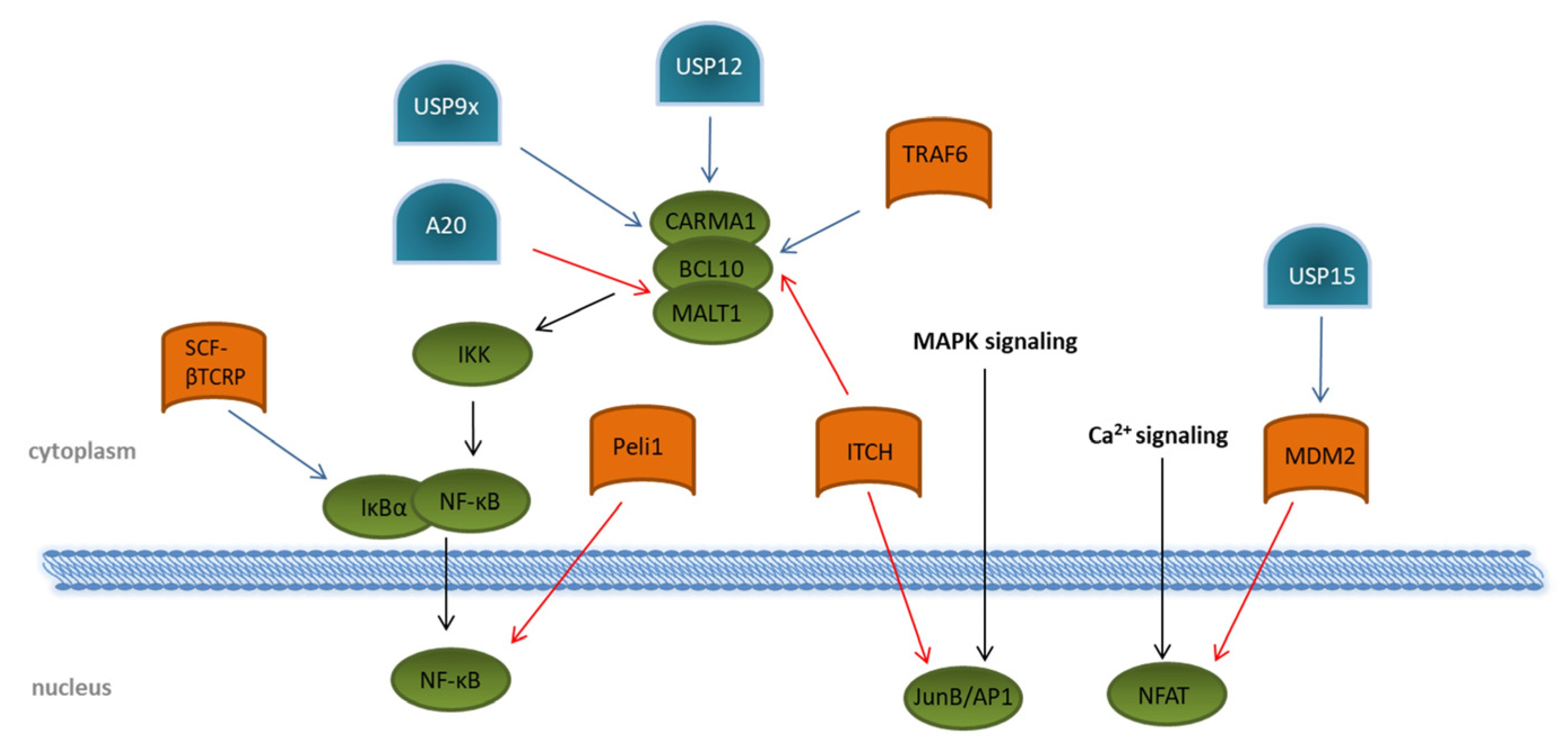
Figure 2). A vast number of transcriptional regulated genes for cytokines, such as IL2, IL6, and IL10; chemokines; and pro-survival proteins, are induced by binding of the NF-κB subunits to the T cell DNA
[23][24]. Activation of NF-κB in TCR signaling is strongly dependent on the formation of the CARMA1–BCL10–MALT1 (CBM) complex, consisting of the CARD-containing MAGUK protein 1 (CARMA1), B-cell lymphoma/leukemia 10 (BCL10), and mucosa-associated lymphoid tissue lymphoma translocation protein 1 homolog (MALT1). This, in turn, activates the IKK complex, resulting in proteasomal degradation of the inhibitory protein IκBα induced by the SCF–βTCRP ubiquitin ligase complex (
Figure 2)
[25][26]. Removal of IκBα allows the active transcription factor NF-κB to translocate into the nucleus
[27][28]. Several E3 ligases are involved in regulating the formation of the CBM complex. ITCH attaches K48 ubiquitin chains to BCL10
[29] for its degradation by proteasomes. The DUBs USP9x, as well as USP12, antagonizes this reaction by specifically hydrolyzing K48-linked ubiquitin chains, thereby supporting the CBM complex association and prolonged the activation of NF-κB
[30][31]. Augmented activation of downstream kinases, such as the inhibitor of nuclear factor-κB (NF-κB) kinases (IKK) α and β, is mediated by the association of the K63 specific E2 enzyme dimer UBC13/UEV1A with the CBM complex. Together with an E3 ligase, K63 ubiquitin chains are then transferred onto BCL10 and MALT1
[32][33]. Although the involved E3 ligase was identified as tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6), a specific TRAF6 knockout revealed no impact on NF-κB activation following TCR triggering, suggesting the contribution of other E3 ligases
[34]. As a counter-regulation, the DUB A20 is able to remove K63-polyubiquitin chains from the CBM complex protein MALT1, thereby downmodulating TCR-induced NF-κB activity
[35]. Interestingly, MALT1 can cleave A20 in a TCR-dependent manner, and this proteolytic cleavage has been suggested to disrupt the ability of A20 to limit TCR downstream signaling via NF-κB inhibition
[36]. A20-dependent downregulation of NF-κB-mediated inflammation is further underlined by the fact that NF-κB activity is negatively regulated by A20 overexpression in mice. In addition, patients with an active autoimmune phenotype due to Behcet’s disease reveal a decreased expression of A20 in CD4+ T cells
[37].
Figure 2. Regulation of transcription-factor activation through ubiquitination and deubiquitination upon T cell stimulation. Overview of known ubiquitin E3 ligases (orange) and deubiquitinating enzymes (blue) involved in the regulation of the transcription factors NF-κB, NFAT, and JunB/AP1 (green). Details are described in the main text of the manuscript. The T cell signaling pathway is indicated by black arrows. Modifications that dampen T cell receptor signaling are indicated by red arrows. Ubiquitin modifications enhancing T cell signaling are indicated by blue arrows.
Direct interaction with the NF-κB subunit c-Rel is mediated by the RING-type E3 ubiquitin protein ligase pellino homolog 1 (PELI1), which attaches K48-linked ubiquitin chains to c-Rel and, thus, initiates its degradation by proteasomes
[38]. Although PELI1 is already constitutively expressed in T cells at a steady state, T cell activation further increases its expression, which subsequently interrupts NF-κB-induced gene transcription, thereby preventing autoimmunity
[38].
Besides NF-κB, members of the NFAT family, including NFATc1, NFATc2, and NFATc4, and the transcription factor JunB/AP1 are activated upon TCR stimulation by increased Ca
2+ flux. Both are responsible for the induced transcription of several cytokines, such as IL2, IL4, TNFα, and IFN-γ. Interestingly, they can cooperatively bind DNA, thereby enhancing the DNA-binding and transcriptional activity of each other
[39][40]. Based on this observation, the question arises whether the regulation of one transcription factor might affect the activity of the other.
With regard to NFAT, the E3 ligase mouse double minute 2 homolog (MDM2) targets the NFATc2 subunit for degradation by ubiquitination
[41]. Interestingly, MDM2 was shown to auto-ubiquitinate itself, leading to K48-linked proteasomal degradation upon T cell activation, which, in turn, amplifies the nuclear accumulation of NFATc2. This is crucial for the induction of cytokines, especially of IFN-γ
[41]. The DUB USP15 antagonizes MDM2 ubiquitination by deubiquitination through a zinc finger motif-mediated mechanism, thereby negatively regulating transcription activity of NFAT and differentiation of IFN-γ-producing Th1 effector T cells (
Figure 4)
[41][42].
Translocation of NFAT to the nucleus is directly influenced by the small ubiquitin-like protein SUMO. The highly sumoylated isoform NFATc1 is translocated to promyelocytic leukemia bodies in the nucleus, leading to the deacetylation of histones and suppression of interleukin-2 gene transcription in vitro
[43]. Recently, NFATc1 sumoylation was analyzed in a transgenic mouse model in which the SUMO modification of NFATc1 was blocked. Accordingly, these mice had significantly high IL2 production and enhanced proliferation of regulatory T cells, as well as suppressed IL17 and IFN-γ release
[44].
The levels of the transcription factor JunB/AP1 depend on ITCH ubiquitination. ITCH-deficient mice had elevated levels of JunB in the nucleus, which led to increased cytokine production and Th2-type inflammation
[45]. Moreover, it has been shown that the sumoylation of JunB is required for the translocation and subsequent expression of T cell-associated cytokine genes like IL2, IL4, and IL10
[44].
Although only a few targets have been determined so far, there is evidence for a regulatory role of neddylation in T cell function. It has been demonstrated that the deficiency of the NEDD8-conjugating enzyme UBC12 leads to diminished proliferation, Th1 and Th2 differentiation, and cytokine production in vitro and in vivo by altered MAPK signaling
[46]. In a parasite infection model, neddylation was required for CD4+ T cell proliferation and Th1-cell differentiation and survival, as well as IFN-γ and anti-parasite IgG responses
[47]. One explanation could be that the neddylation of cullin-ring ligases is responsible for the degradation of IκBα, thereby influencing the NF-κB pathway. The pharmacological blockade of the entire neddylation by MLN4924/Pevonedistad has been proven to cause an accumulation of IκBα and abrogated translocation of NF-κB into the nucleus in T cells from chronic lymphocytic leukemia
[22]. Therefore, it is very likely that the observed effects are due to changes in NF-κB translocation. However, the exact targets have not yet been identified.
Thus, T cell-specific transcription factors fulfill multiple redundant functions, with the disruption of one factor affecting the function of the other. Ubiquitination, sumoylation, and, in part, neddylation are critical processes for regulating transcription factor trafficking and effector functions.
3. T Cell Development
Mature T cells are constantly recirculating between peripheral lymphoid organs and the bloodstream until they encounter their specific antigen. To ensure that T cells respond to foreign antigens from invading pathogens and simultaneously tolerate self-antigens, T cells have to be educated during thymic development. In the thymus, positive and negative selection processes define the assembled T cell repertoire that only T cell receptors with a weak affinity for self-antigenic peptides bound to the major histocompatibility complex (MHC) are selected. The development and selection of T cells is provided by thymic epithelial cells (TECs) divided in cortical TECs responsible for positive selection and medullary TECs (mTECs) crucial for negative selection
[48]. Therefore, mTEC maturation is a critical step in thymic selection, which is regulated by activation of the NF-κB signaling pathway via the TNF receptor superfamily members CD40, receptor activator of NF-κB (RANK), and lymphotoxin β receptor (LTβR)
[49][50][51]. Studies on TRAF6 knockout mice, an essential mediator in NF-κB activation that displays E3 ligase activity, have been accompanied with the expression of fewer tissue-specific antigens and compromised mTEC development, leading to an autoimmune phenotype
[52]. Moreover, TRAF6 deficiency results in a reduced expression of the autoimmune regulator (AIRE), which is pivotal for negative selection mediating the expression of many tissue-restricted self-antigens. AIRE consists of two plant homeodomains (PHDs), in which PHD1 is speculated to have E3 ligase activity
[53]. In the last twenty years, numerous functions of AIRE have been described. Due to its nuclear localization, it binds chromatin and histones and is involved in gene transcription. Furthermore, it is able to enhance alternative mRNA splicing. Altogether, this enables AIRE to regulate the expression of tissue-specific antigens in the mTECs mandatory for negative selection
[54].
In addition, the ubiquitin-like protein FAT10 contributes to negative selection. FAT 10 has been shown to be expressed in mTECs
[55]. The knockout of FAT10 in mice revealed changes in the T cell repertoire of CD4 and CD8 single positive cells during thymic development, probably due to an altered peptide presentation that influences negative selection
[56].
In summary, the regulation of T cell selection processes is crucial for maintaining a functional adaptive immune response. Mutations in specific E3 ligases or DUBs critically affect T cell homeostasis and often lead to alterations in immune responses that ultimately can result in autoimmune diseases and cancer, as well as complications in fighting infectious diseases.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23073424