Innate lymphoid cells (ILCs) represent a heterogeneous population of non-B/non-T lymphocytes whose discovery has greatly expanded researchers understanding over the past 10 years. ILCs are defined mainly by three unique features: (1) their lymphoid morphology; (2) their lack of genetically rearranged antigen receptors; and (3) their deficiency of cell-surface markers expressed in other immune cell types, such as myeloid cells and dendritic cells. In recent years, emerging evidence has identified the existence of ILCs in the pancreas, and all ILC subsets have been identified. ILCs resident in the pancreas, as well as in other tissues, may play an important role in the occurrence and progression of pancreatic diseases and will therefore be expected to be applied in the prevention and treatment of these diseases.
- innate lymphoid cells
- diabetes mellitus
- pancreatitis
- pancreatic cancer
1. Introduction
In recent years, emerging evidence has identified the existence of ILCs in the pancreas, and all ILC subsets have been identified. ILCs resident in the pancreas, as well as in other tissues, may play an important role in the occurrence and progression of pancreatic diseases and will therefore be expected to be applied in the prevention and treatment of these diseases.
2. A Fundamental Overview of ILCs: Phenotype and Functions
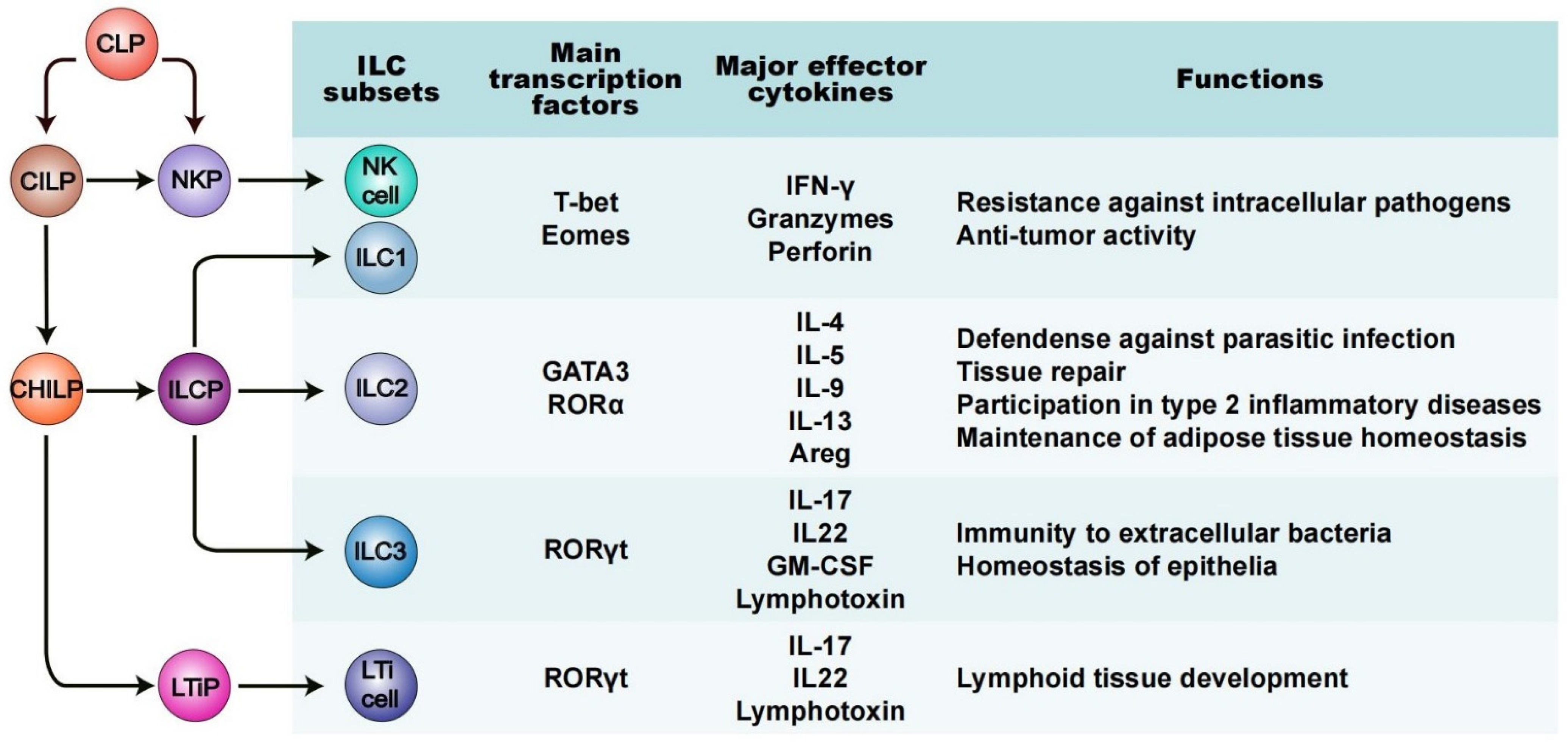
3. ILCs in Diabetes Mellitus
Diabetes mellitus is a progressive and complex metabolic disorder, characterized by chronic hyperglycemia, caused by impaired insulin secretion and (or) utilization.
ILCs resident in AT have been proven to limit or promote the development of obesity and obesity-associated T2DM (Figure 2). In healthy lean individuals, AT is enriched with type 2 immune cells, such as ILC2s, eosinophils and alternatively activated macrophages (AAMs, anti-inflammatory or M2 macrophages), to maintain tissue homeostasis and support a metabolically healthy state. During the process of type 2 immunity in AT, ILC2 plays an integral role in communication and regulation, and are therefore indispensable regulators. On the one hand, ILC2s produce cytokines IL-5 and IL-13, promoting the recruitment and accumulation of eosinophils and AAMs to support AT remodeling and to restrict “type 1” inflammatory responses. On the other hand, ILC2s promote the beiging of white adipose tissue (WAT), contributing to an increasing of both the quantity and the performance of beige adipocytes in AT. Through the two known mechanisms above, ILC2s help with the maintenance of AT balance and protect from obesity-associated metabolic dysfunction, insulin resistance and T2DM. In the AT of obese patients, diet-induced obesity initiates the early production of IL-12, which results in selective proliferation and accumulation of ILC1s which requires the IL-12 receptor and STAT4 signaling. ILC1-derived IFN-γ is necessary to accelerate classically activated macrophages (CAMs, proinflammatory macrophages or M1 macrophages) polarization and contributes to obesity-associated insulin resistance. In addition, adipose ILC1s have also been demonstrated to promote AT fibrogenesis by increasing M1 macrophages and activating the TGF-β1/Smad3 signaling pathway. By recruiting and activating M1 macrophages and inducing AT fibrosis, adipose-resident ILC1s participate in and promote the progression of insulin resistance and obesity-associated diabetes.
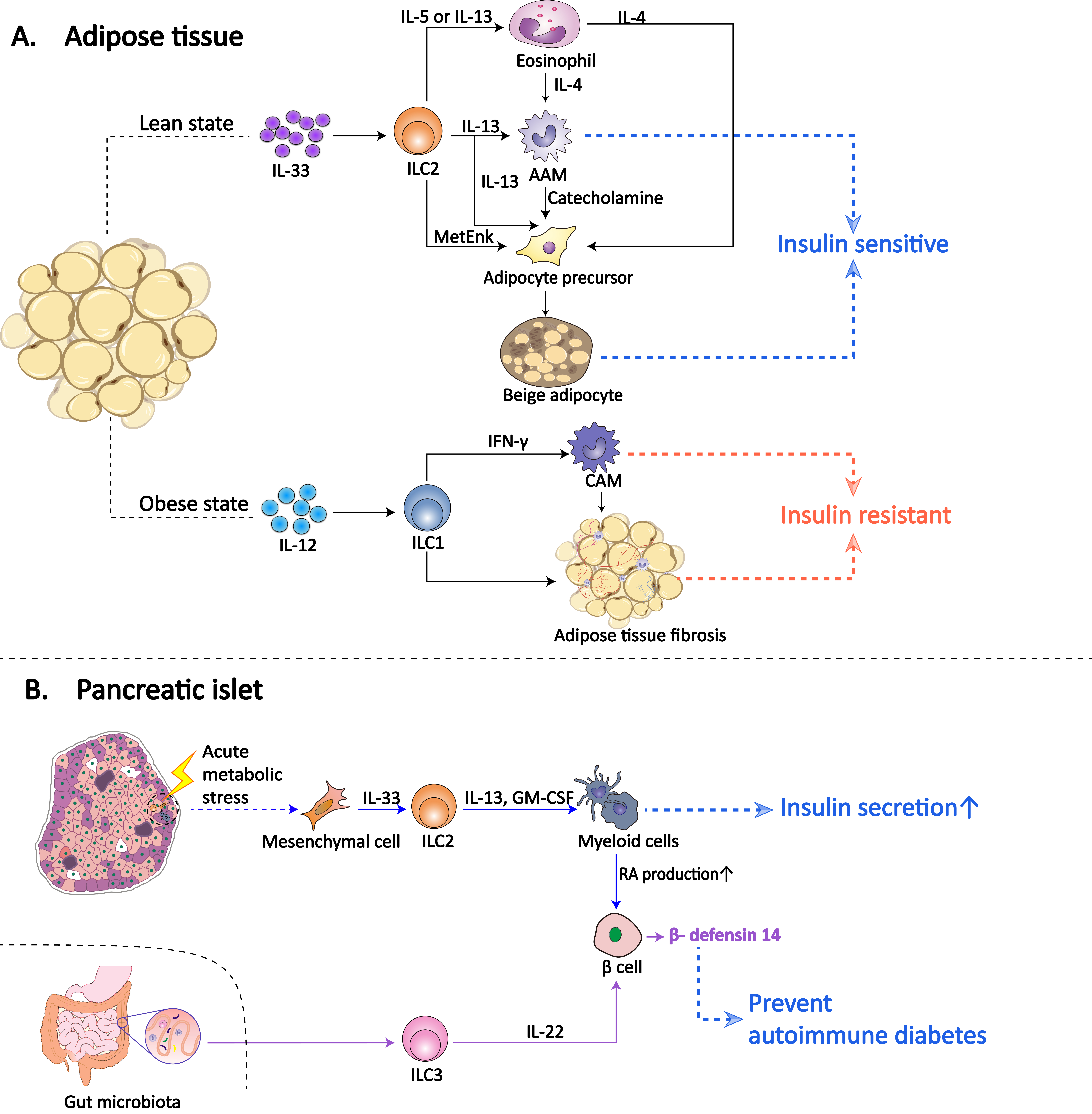
Figure 2. Role of ILCs in diabetes mellitus. A | ILCs in adipose tissue. In the lean state, IL-33 induces adipose-resident ILC2s to produce the cytokines IL-5 or IL-13, which support the recruitment and accumulation of eosinophils in AT. Eosinophils produce IL-4 to sustain and recruit AAMs. ILC2s produce ample IL 13 and may also directly contribute to AAM recruitment and maintenance. AAM byproducts, such as IL-10, contribute to adipocyte insulin sensitivity and protect against DM. In addition, IL-4, IL-13 and methionine-enkephalin peptides (MetEnk) and catecholamines, produced by eosinophils, ILC2s and AAMs, respectively, promote the proliferation and differentiation of adipocyte precursors into beige adipocytes. Beige fat biogenesis also promotes insulin sensitivity and prevents DM. In the obese state, while IL-12 promotes the selective accumulation of adipose-resident ILC1s. ILC1s drive CAM polarization by IFN-γ production and promote AT fibrosis, contributing to obesity-associated insulin resistance and DM. B | ILCs in pancreatic islets. In diabetic or obese states, the islets are also in an inflammatory background. IL-33 is produced by mesenchymal cells as a stress signal in islets. As the main IL-33-responsive cells in islets, islet-resident ILC2s stimulate the capacity of myeloid cells to produce RA, which in turn enhances insulin secretion in islet β cells and protects against DM. In the gut, the microbiota controls IL-22 expression by ILC3s within pancreatic islets through different pathways. ILC3-derived IL-22 induces islet β cells to produce β-defensin, preventing autoimmune diabetes.
A variety of substances that constitute the diabetic milieu, such as glucose and saturated fatty acids, stimulate the islet to produce various proinflammatory chemokines and cytokines and recruit and activate type 1 immune cells. Therefore, anti-inflammatory drugs for T2DM treatment are under development. Using immunofluorescence analyses, Dalmas et al. confirmed the existence of ILC2s located inside or in the periphery of islets in the mouse pancreas. Under conditions of islet inflammation in T2DM, proinflammatory factors induce mesenchymal cell-derived IL-33. Islet-resident ILC2s expressing the IL-33 receptor (IL-33R) are the major IL-33-responsive cells in islets. ILC2s increase the number of islet myeloid cells and elicit their capacity to promote retinoic acid (RA) production in a manner dependent on the secretion of IL-13 and GM-CSF. Ultimately, increased RA in turn enhances insulin secretion in islet β cells. The process above may be associated in part with the phenotypic plasticity shown by ILC2s in response to inflammatory signaling.
Islet-resident ILC3s have been found to play a role in protecting against autoimmune diabetes in mouse models (Figure 2). This function is mainly achieved by ILC3-induced mouse β-defensin 14 (mBD14) expression, and the activation of the former depends on gut microbiota. Defensin is a kind of antimicrobial peptide whose abnormal expression has been proven to be associated with diseases, including autoimmune diabetes. In the gut, the microbiota is known to control IL-22 expression in ILC3s through different pathways. On the one hand, by expressing aryl hydrocarbon receptor (AHR) ligands, some specific gut microbes can directly stimulate ILC3s to produce IL-22. On the other hand, other gut microbiota indirectly positively affects ILC3s via the induction of IL-23 secretion (a strong inducer of IL-22) by intestinal phagocytes. These pathways also work in islets to stimulate islet-resident ILC3s to secrete IL-22. In pancreatic islet, ILC3s are the major source of IL-22. ILC3-derived IL-22 induces islet β cells to produce mBD14, preventing autoimmune diabetes through the ILC3-IL22-mBD14 axis. In addition, ILC3s have also been found to produce GM-CSF and thus may play a partial role in regulating insulin secretion and protecting against T2DM.
4. ILCs (Mainly NK Cells) in Pancreatitis
Pancreatitis is an inflammatory disease of pancreatic tissue. Different etiologies, including pancreatic duct obstruction secondary to gallstones, alcohol abuse, as well as surgical trauma or pharmacological means, cause the dysfunction of cellular pathways and organelles, ultimately leading to acinar cell death and local and systemic inflammation. As a recently discovered immune cell group, the role of ILCs in pancreatitis has not been well studied. Current studies are mainly aimed at NK cells, but the knowledge gained is still relatively limited.
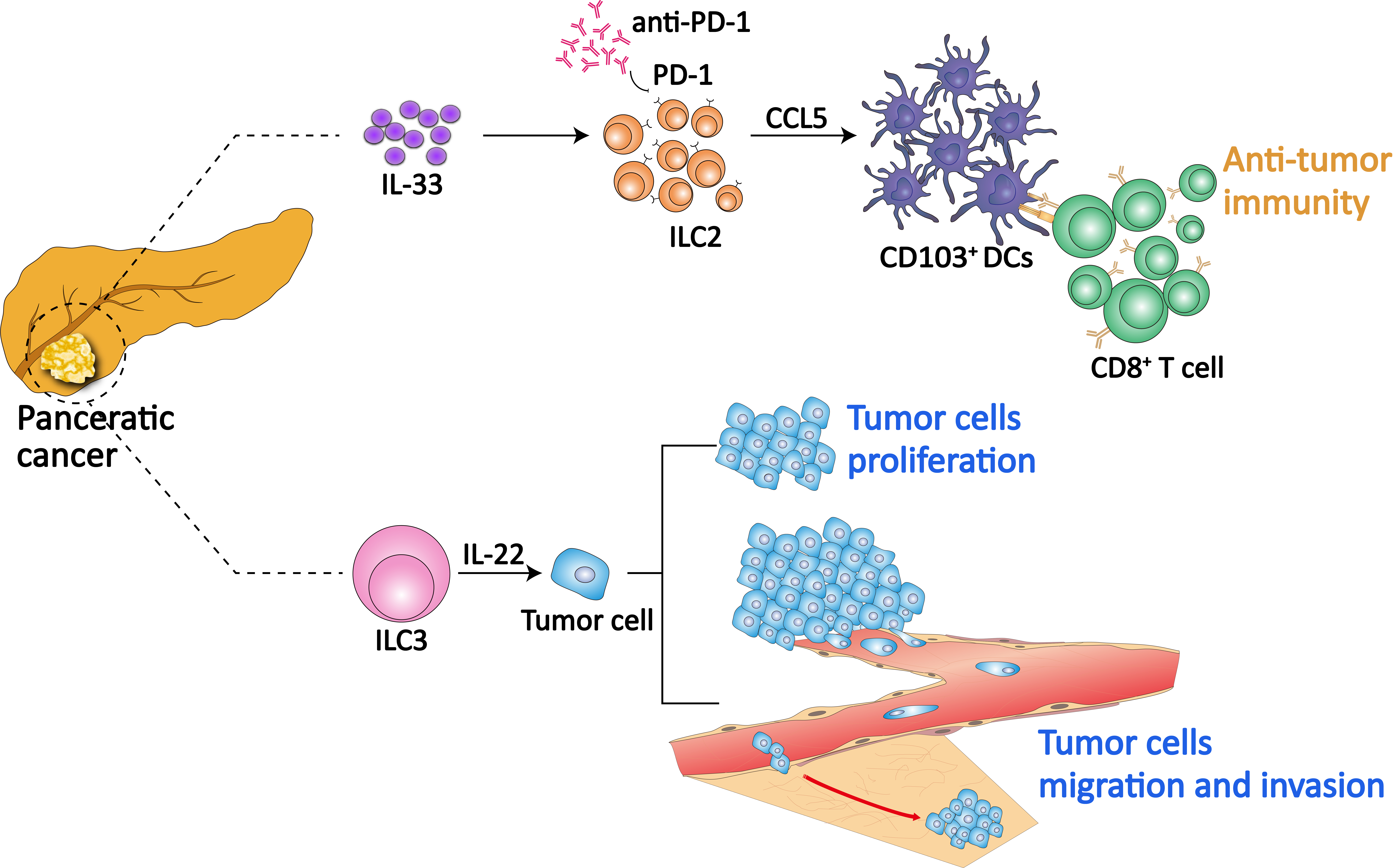
5. ILCs in Pancreatic Cancer
This entry is adapted from the peer-reviewed paper 10.3390/ijms23073748
References
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149.
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells: 10 years on. Cell 2018, 174, 1054–1066.
- Gasteiger, G.; Fan, X.; Dikiy, S.; Lee, S.Y.; Rudensky, A.Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 2015, 350, 981–985.
- Kim, C.H.; Hashimoto-Hill, S.; Kim, M. Migration and tissue tropism of innate lymphoid cells. Trends Immunol. 2016, 37, 68–79.
- Meininger, I.; Carrasco, A.; Rao, A.; Soini, T.; Kokkinou, E.; Mjosberg, J. Tissue-specific features of innate lymphoid cells. Trends Immunol. 2020, 41, 902–917.
- Kobayashi, T.; Ricardo-Gonzalez, R.R.; Moro, K. Skin-resident innate lymphoid cells—Cutaneous innate guardians and regulators. Trends Immunol. 2020, 41, 100–112.
- Pearson, C.; Uhlig, H.H.; Powrie, F. Lymphoid microenvironments and innate lymphoid cells in the gut. Trends Immunol. 2012, 33, 289–296.
- Diefenbach, A.; Gnafakis, S.; Shomrat, O. Innate lymphoid cell-epithelial cell modules sustain intestinal homeostasis. Immunity 2020, 52, 452–463.
- Barlow, J.L.; McKenzie, A.N.J. Innate lymphoid cells of the lung. Annu. Rev. Physiol. 2019, 81, 429–452.
- Eberl, G.; Colonna, M.; di Santo, J.P.; McKenzie, A.N. Innate lymphoid cells. Innate lymphoid cells: A new paradigm in immunology. Science 2015, 348, aaa6566.
- Klose, C.S.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774.
- Branzk, N.; Gronke, K.; Diefenbach, A. Innate lymphoid cells, mediators of tissue homeostasis, adaptation and disease tolerance. Immunol. Rev. 2018, 286, 86–101.
- Sonnenberg, G.F.; Hepworth, M.R. Functional interactions between innate lymphoid cells and adaptive immunity. Nat. Rev. Immunol. 2019, 19, 599–613.
- Bénézech, C.; Jackson-Jones, L.H. ILC2 orchestration of local immune function in adipose tissue. Front. Immunol. 2019, 10, 171.
- Wang, R.; Wang, Y.; Harris, D.C.H.; Cao, Q. Innate lymphoid cells in kidney diseases. Kidney Int. 2021, 99, 1077–1087.
- Forkel, M.; Berglin, L.; Kekäläinen, E.; Carlsson, A.; Svedin, E.; Michaëlsson, J.; Nagasawa, M.; Erjefält, J.S.; Mori, M.; Flodström-Tullberg, M.; et al. Composition and functionality of the intrahepatic innate lymphoid cell-compartment in human nonfibrotic and fibrotic livers. Eur. J. Immunol. 2017, 47, 1280–1294.
- Cuff, A.O.; Sillito, F.; Dertschnig, S.; Hall, A.; Luong, T.V.; Chakraverty, R.; Male, V. The obese liver environment mediates conversion of NK cells to a less cytotoxic ILC1-like phenotype. Front. Immunol. 2019, 10, 2180.
- von Burg, N.; Turchinovich, G.; Finke, D. Maintenance of immune homeostasis through ILC/T cell interactions. Front. Immunol. 2015, 6, 416.
- Withers, D.R. Innate lymphoid cell regulation of adaptive immunity. Immunology 2016, 149, 123–130.
- Ebbo, M.; Crinier, A.; Vely, F.; Vivier, E. Innate lymphoid cells: Major players in inflammatory diseases. Nat. Rev. Immunol. 2017, 17, 665–678.
- Bartemes, K.R.; Kita, H. Roles of innate lymphoid cells (ILCs) in allergic diseases: The 10-year anniversary for ILC2s. J. Allergy Clin. Immunol. 2021, 147, 1531–1547.
- Galy, A.; Travis, M.; Cen, D.; Chen, B. Human T, B, natural killer, and dendritic cells arise from a common bone marrow progenitor cell subset. Immunity 1995, 3, 459–473.
- Kondo, M.; Weissman, I.L.; Akashi, K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 1997, 91, 661–672.
- Guia, S.; Narni-Mancinelli, E. Helper-like innate lymphoid cells in humans and mice. Trends Immunol. 2020, 41, 436–452.
- Cherrier, D.E.; Serafini, N.; Di Santo, J.P. Innate lymphoid cell development: A T cell perspective. Immunity 2018, 48, 1091–1103.
- Colonna, M. Innate lymphoid cells: Diversity, plasticity, and unique functions in immunity. Immunity 2018, 48, 1104–1117.
- Spits, H.; Di Santo, J.P. The expanding family of innate lymphoid cells: Regulators and effectors of immunity and tissue remodeling. Nat. Immunol. 2011, 12, 21–27.
- Koues, O.I.; Collins, P.L.; Cella, M.; Robinette, M.L.; Porter, S.I.; Pyfrom, S.C.; Payton, J.E.; Colonna, M.; Oltz, E.M. Distinct gene regulatory pathways for human innate versus adaptive lymphoid cells. Cell 2016, 165, 1134–1146.
- Ercolano, G.; Wyss, T.; Salomé, B.; Romero, P.; Trabanelli, S.; Jandus, C. Distinct and shared gene expression for human innate versus adaptive helper lymphoid cells. J. Leukoc. Biol. 2020, 108, 723–737.
- Kiessling, R.; Klein, E.; Pross, H.; Wigzell, H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 1975, 5, 117–121.
- Daussy, C.; Faure, F.; Mayol, K.; Viel, S.; Gasteiger, G.; Charrier, E.; Bienvenu, J.; Henry, T.; Debien, E.; Hasan, U.A.; et al. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J. Exp. Med. 2014, 211, 563–577.
- Gordon, S.M.; Chaix, J.; Rupp, L.J.; Wu, J.; Madera, S.; Sun, J.C.; Lindsten, T.; Reiner, S.L. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 2012, 36, 55–67.
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469.
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49.
- Abt, M.C.; Lewis, B.B.; Caballero, S.; Xiong, H.; Carter, R.A.; Sušac, B.; Ling, L.; Leiner, I.; Pamer, E.G. Innate immune defenses mediated by two ILC subsets are critical for protection against acute clostridium difficile infection. Cell Host Microbe 2015, 18, 27–37.
- Weizman, O.-E.; Adams, N.M.; Schuster, I.S.; Krishna, C.; Pritykin, Y.; Lau, C.; Degli-Esposti, M.A.; Leslie, C.S.; Sun, J.C.; O’Sullivan, T.E. ILC1 confer early host protection at initial sites of viral infection. Cell 2017, 171, 795–808.
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity 2013, 38, 769–781.
- Zhang, J.; Marotel, M.; Fauteux-Daniel, S.; Mathieu, A.-L.; Viel, S.; Marçais, A.; Walzer, T. T-bet and Eomes govern differentiation and function of mouse and human NK cells and ILC1. Eur. J. Immunol. 2018, 48, 738–750.
- Cortez, V.S.; Colonna, M. Diversity and function of group 1 innate lymphoid cells. Immunol. Lett. 2016, 179, 19–24.
- Yagi, R.; Zhong, C.; Northrup, D.L.; Yu, F.; Bouladoux, N.; Spencer, S.; Hu, G.; Barron, L.; Sharma, S.; Nakayama, T.; et al. The transcription factor GATA3 is critical for the development of all IL-7Rα-expressing innate lymphoid cells. Immunity 2014, 40, 378–388.
- Wong, S.H.; Walker, J.A.; Jolin, H.E.; Drynan, L.F.; Hams, E.; Camelo, A.; Barlow, J.L.; Neill, D.R.; Panova, V.; Koch, U.; et al. Transcription factor RORα is critical for nuocyte development. Nat. Immunol. 2012, 13, 229–236.
- Fallon, P.G.; Ballantyne, S.J.; Mangan, N.E.; Barlow, J.L.; Dasvarma, A.; Hewett, D.R.; McIlgorm, A.; Jolin, H.E.; McKenzie, A.N.J. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J. Exp. Med. 2006, 203, 1105–1116.
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.-I.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of TH2 cytokines by adipose tissue-associated c-Kit+Sca-1+ lymphoid cells. Nature 2010, 463, 540–544.
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.A.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370.
- Price, A.E.; Liang, H.-E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11489–11494.
- Cella, M.; Fuchs, A.; Vermi, W.; Facchetti, F.; Otero, K.; Lennerz, J.K.M.; Doherty, J.M.; Mills, J.C.; Colonna, M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009, 457, 722–725.
- Wang, Y.; Koroleva, E.P.; Kruglov, A.A.; Kuprash, D.V.; Nedospasov, S.A.; Fu, Y.-X.; Tumanov, A.V. Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity 2010, 32, 403–413.
- Cupedo, T.; Crellin, N.K.; Papazian, N.; Rombouts, E.J.; Weijer, K.; Grogan, J.L.; Fibbe, W.E.; Cornelissen, J.J.; Spits, H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol. 2009, 10, 66–74.
- Eberl, G.; Marmon, S.; Sunshine, M.-J.; Rennert, P.D.; Choi, Y.; Littman, D.R. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol. 2004, 5, 64–73.
- Mebius, R.E.; Rennert, P.; Weissman, I.L. Developing lymph nodes collect CD4+CD3− LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 1997, 7, 493–504.
- Yoshida, H.; Honda, K.; Shinkura, R.; Adachi, S.; Nishikawa, S.; Maki, K.; Ikuta, K.; Nishikawa, S.I. IL-7 receptor α+ CD3– cells in the embryonic intestine induces the organizing center of Peyer’s patches. Int. Immunol. 1999, 11, 643–655.
- Fraker, C.; Bayer, A.L. The expanding role of natural killer cells in type 1 diabetes and immunotherapy. Curr. Diabetes Rep. 2016, 16, 109.
- Marca, V.; Gianchecchi, E.; Fierabracci, A. Type 1 diabetes and its multi-factorial pathogenesis: The putative role of NK cells. Int. J. Mol. Sci. 2018, 19, 794.
- Jewett, A.; Kos, J.; Fong, Y.; Ko, M.-W.; Safaei, T.; Nanut, M.P.; Kaur, K. NK cells shape pancreatic and oral tumor microenvironments; role in inhibition of tumor growth and metastasis. Semin. Cancer Biol. 2018, 53, 178–188.
- Peng, X.; Chen, L.; Jiao, Y.; Wang, Y.; Hao, Z.; Zhan, X. Application of natural killer cells in pancreatic cancer. Oncol. Lett. 2021, 22, 647.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics 2020. CA Cancer J. Clin. 2020, 70, 7–30.
- Gürlevik, E.; Fleischmann-Mundt, B.; Brooks, J.; Demir, I.E.; Steiger, K.; Ribback, S.; Yevsa, T.; Woller, N.; Kloos, A.; Ostroumov, D.; et al. Administration of gemcitabine after pancreatic tumor resection in mice induces an antitumor immune response mediated by natural killer cells. Gastroenterology 2016, 151, 338–350.
- Jewett, A.; Kos, J.; Kaur, K.; Safaei, T.; Sutanto, C.; Chen, W.; Wong, P.; Namagerdi, A.K.; Fang, C.; Fong, Y.; et al. Natural killer cells: Diverse functions in tumor immunity and defects in pre-neoplastic and neoplastic stages of tumorigenesis. Mol. Ther. Oncolytics 2020, 16, 41–52.
- Moral, J.A.; Leung, J.; Rojas, L.A.; Ruan, J.; Zhao, J.; Sethna, Z.; Ramnarain, A.; Gasmi, B.; Gururajan, M.; Redmond, D.; et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature 2020, 579, 130–135.
- Thompson, R.H.; Dong, H.; Lohse, C.M.; Leibovich, B.C.; Blute, M.L.; Cheville, J.C.; Kwon, E.D. PD-1 is expressed by tumor-infiltrating immune cells and is associated with poor outcome for patients with renal cell carcinoma. Clin. Cancer Res. 2007, 13, 1757–1761.
- Shi, F.; Shi, M.; Zeng, Z.; Qi, R.-Z.; Liu, Z.-W.; Zhang, J.-Y.; Yang, Y.-P.; Tien, P.; Wang, F.-S. PD-1 and PD-L1 upregulation promotes CD8+ T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer 2011, 128, 887–896.
- French, J.D.; Kotnis, G.R.; Said, S.; Raeburn, C.D.; McIntyre, R.C.; Klopper, J.P.; Haugen, B.R. Programmed death-1+ T cells and regulatory T cells are enriched in tumor-involved lymph nodes and associated with aggressive features in papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2012, 97, E934–E943.
- Sun, S.; Fei, X.; Mao, Y.; Wang, X.; Garfield, D.H.; Huang, O.; Wang, J.; Yuan, F.; Sun, L.; Yu, Q.; et al. PD-1+ immune cell infiltration inversely correlates with survival of operable breast cancer patients. Cancer Immunol. Immunother. 2014, 63, 395–406.
- Yu, Y.; Tsang, J.C.H.; Wang, C.; Clare, S.; Wang, J.; Chen, X.; Brandt, C.; Kane, L.; Campos, L.S.; Lu, L.; et al. Single-cell RNA-seq identifies a PD-1 ILC progenitor and defines its development pathway. Nature 2016, 539, 102–106.
- Taylor, S.; Huang, Y.; Mallett, G.; Stathopoulou, C.; Felizardo, T.C.; Sun, M.-A.; Martin, E.L.; Zhu, N.; Woodward, E.L.; Elias, M.S.; et al. PD-1 regulates KLRG1 group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1663–1678.
- Araujo, J.M.; Gomez, A.C.; Aguilar, A.; Salgado, R.; Balko, J.M.; Bravo, L.; Doimi, F.; Bretel, D.; Morante, Z.; Flores, C.; et al. Effect of CCL5 expression in the recruitment of immune cells in triple negative breast cancer. Sci. Rep. 2018, 8, 4899.
- Ikutani, M.; Yanagibashi, T.; Ogasawara, M.; Tsuneyama, K.; Yamamoto, S.; Hattori, Y.; Kouro, T.; Itakura, A.; Nagai, Y.; Takaki, S.; et al. Identification of innate IL-5-producing cells and their role in lung eosinophil regulation and antitumor immunity. J. Immunol. 2012, 188, 703–713.
- Saranchova, I.; Han, J.; Zaman, R.; Arora, H.; Huang, H.; Fenninger, F.; Choi, K.B.; Munro, L.; Pfeifer, C.G.; Welch, I.; et al. Type 2 innate lymphocytes actuate immunity against tumours and limit cancer metastasis. Sci. Rep. 2018, 8, 2924.
- Xuan, X.; Zhou, J.; Tian, Z.; Lin, Y.; Song, J.; Ruan, Z.; Ni, B.; Zhao, H.; Yang, W. ILC3 cells promote the proliferation and invasion of pancreatic cancer cells through IL-22/AKT signaling. Clin. Transl. Oncol. 2020, 22, 563–575.
- Carrega, P.; Loiacono, F.; di Carlo, E.; Scaramuccia, A.; Mora, M.; Conte, R.; Benelli, R.; Spaggiari, G.M.; Cantoni, C.; Campana, S.; et al. NCR+ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat. Commun. 2015, 6, 8280.
- Saalim, M.; Resham, S.; Manzoor, S.; Ahmad, H.; Jaleel, S.; Ashraf, J.; Imran, M.; Naseem, S. IL-22: A promising candidate to inhibit viral-induced liver disease progression and hepatocellular carcinoma. Tumour. Biol. 2016, 37, 105–114.
- Zhuang, Y.; Peng, L.-S.; Zhao, Y.-L.; Shi, Y.; Mao, X.-H.; Guo, G.; Chen, W.; Liu, X.-F.; Zhang, J.-Y.; Liu, T.; et al. Increased intratumoral IL-22-producing CD4+ T cells and Th22 cells correlate with gastric cancer progression and predict poor patient survival. Cancer Immunol. Immunother. 2012, 61, 1965–1975.
- Kirchberger, S.; Royston, D.J.; Boulard, O.; Thornton, E.; Franchini, F.; Szabady, R.L.; Harrison, O.; Powrie, F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 2013, 210, 917–931.
- Tugues, S.; Ducimetiere, L.; Friebel, E.; Becher, B. Innate lymphoid cells as regulators of the tumor microenvironment. Semin. Immunol. 2019, 41, 101270.