Phenotypical screening is a widely used approach in drug discovery for the identification of small molecules with cellular activities. However, functional annotation of identified hits often poses a challenge. The development of small molecules with narrow or exclusive target selectivity such as chemical probes and chemogenomic (CG) libraries, greatly diminishes this challenge, but non-specific effects caused by compound toxicity or interference with basic cellular functions still pose a problem to associate phenotypic readouts with molecular targets. Hence, each compound should ideally be comprehensively characterized regarding its effects on general cell functions. Here, we report an optimized live-cell multiplexed assay that classifies cells based on nuclear morphology, presenting an excellent indicator for cellular responses such as early apoptosis and necrosis. This basic readout, in combination with the detection of other general cell damaging activities of small molecules such as changes in cytoskeletal morphology, cell cycle and mitochondrial health, provides a comprehensive time-dependent characterization of the effect of small molecules on cellular health in a single experiment. The developed high-content assay offers multi-dimensional comprehensive characterization that can be used to delineate generic effects regarding cell functions and cell viability, allowing an assessment of compound suitability for subsequent detailed phenotypic and mechanistic studies.
- phenotypic screening
- high content imaging
- chemogenomics
- machine learning
- cell cycle
1. Introduction
2. Image-Based Annotation of Chemogenomic Libraries for Phenotypic Screening
2.2. Multiplex Protocol
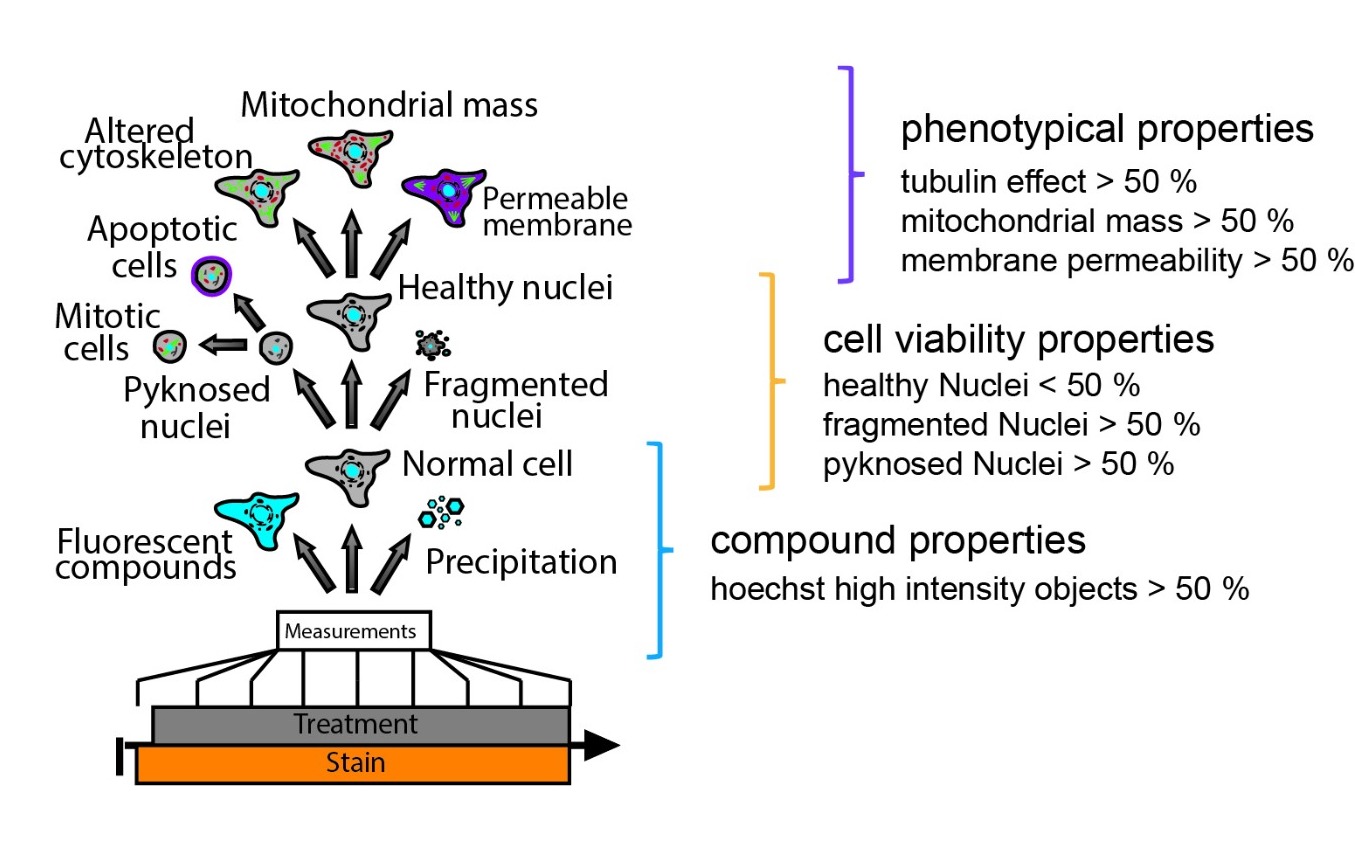
Figure 1.: General workflow of Multiplex High Via protocol analysis with property thresholds.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27041439
References
- Haasen, D.; Schopfer, U.; Antczak, C.; Guy, C.; Fuchs, F.; Selzer, P. How Phenotypic Screening Influenced Drug Discovery: Lessons from Five Years of Practice. ASSAY Drug Dev. Technol. 2017, 15, 239–246.
- Rietdijk, J.; Tampere, M.; Pettke, A.; Georgiev, P.; Lapins, M.; Warpman-Berglund, U.; Spjuth, O.; Puumalainen, M.-R.; Carreras-Puigvert, J. A phenomics approach for antiviral drug discovery. BMC Biol. 2021, 19, 156.
- Bray, M.-A.; Singh, S.; Han, H.; Davis, C.T.; Borgeson, B.; Hartland, C.; Kost-Alimova, M.; Gustafsdottir, S.M.; Gibson, C.C.; Carpenter, A. Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat. Protoc. 2016, 11, 1757–1774.
- Schiff, L.; Migliori, B.; Chen, Y.; Carter, D. Deep learning and automated Cell Painting reveal Parkinson’s disease-specific signatures in primary patient fibroblasts. bioRxiv 2020.
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543.
- Arrowsmith, C.; Audia, J.; Austin, C.; Baell, J.; Bennett, J.; Blagg, J.; Bountra, C.; Brennan, P.; Brown, P.; Bunnage, M.E.; et al. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 536–541.
- Brown, P.J.; Müller, S. Open access chemical probes for epigenetic targets. Futur. Med. Chem. 2015, 7, 1901–1917.
- Drewes, G.; Knapp, S. Chemoproteomics and Chemical Probes for Target Discovery. Trends Biotechnol. 2018, 36, 1275–1286.
- Bunnage, M.E.; Chekler, E.L.P.; Jones, L. Target validation using chemical probes. Nat. Chem. Biol. 2013, 9, 195–199.
- Wells, C.I.; Al-Ali, H.; Andrews, D.M.; Asquith, C.R.M.; Axtman, A.D.; Dikic, I.; Ebner, D.; Ettmayer, P.; Fischer, C.; Frederiksen, M.; et al. The Kinase Chemogenomic Set (KCGS): An Open Science Resource for Kinase Vulnerability Identification. Int. J. Mol. Sci. 2021, 22, 566.
- Canham, S.M.; Wang, Y.; Cornett, A.; Auld, D.S.; Baeschlin, D.K.; Patoor, M.; Skaanderup, P.R.; Honda, A.; Llamas, L.; Wendel, G.; et al. Systematic Chemogenetic Library Assembly. Cell Chem. Biol. 2020, 27, 1124–1129.
- Dafniet, B.; Cerisier, N.; Boezio, B.; Clary, A.; Ducrot, P.; Dorval, T.; Gohier, A.; Brown, D.; Audouze, K.; Taboureau, O. Development of a chemogenomics library for phenotypic screening. J. Chemin. 2021, 13, 91.
- Müller, S.; Ackloo, S.; Arrowsmith, C.H.; Bauser, M.; Baryza, J.L.; Blagg, J.; Boettcher, J.; Bountra, C.; Brown, P.; Bunnage, M.; et al. Donated chemical probes for open science. eLife 2018, 7, 7.
- Bredel, M.; Jacoby, E. Chemogenomics: An emerging strategy for rapid target and drug discovery. Nat. Rev. Genet. 2004, 5, 262–275.
- Jones, L.; Bunnage, M.E. Applications of chemogenomic library screening in drug discovery. Nat. Rev. Drug Discov. 2017, 16, 285–296.
- Caron, P.R.; Mullican, M.D.; Mashal, R.D.; Wilson, K.P.; Su, M.S.; Murcko, M. Chemogenomic approaches to drug discovery. Curr. Opin. Chem. Biol. 2001, 5, 464–470.
- >EUbOPEN. Available online: https://www.eubopen.org/ (accessed on 5 January 2022).
- Carter, A.J.; Kraemer, O.; Zwick, M.; Mueller-Fahrnow, A.; Arrowsmith, C.H.; Edwards, A.M. Target 2035: Probing the human proteome. Drug Discov. Today 2019, 24, 2111–2115.
- Kawamura, T.; Kawatani, M.; Muroi, M.; Kondoh, Y.; Futamura, Y.; Aono, H.; Tanaka, M.; Honda, K.; Osada, H. Proteomic profiling of small-molecule inhibitors reveals dispensability of MTH1 for cancer cell survival. Sci. Rep. 2016, 6, 26521.
- Sun, H.; Wang, Y.; Cheff, D.M.; Hall, M.D.; Shen, M. Predictive models for estimating cytotoxicity on the basis of chemical structures. Bioorg. Med. Chem. 2020, 28, 115422.
- Tang, H.; Duggan, S.; Richardson, P.L.; Marin, V.; Warder, S.E.; McLoughlin, S.M. Target Identification of Compounds from a Cell Viability Phenotypic Screen Using a Bead/Lysate-Based Affinity Capture Platform. J. Biomol. Screen. 2015, 21, 201–211.
- Boutros, M.; Heigwer, F.; Laufer, C. Microscopy-Based High-Content Screening. Cell 2015, 163, 1314–1325.
- Chandrasekaran, S.N.; Ceulemans, H.; Boyd, J.D.; Carpenter, A.E. Image-based profiling for drug discovery: Due for a machine-learning upgrade? Nat. Rev. Drug Discov. 2021, 20, 145–159.
- Cole, R. Live-cell imaging. Cell Adhes. Migr. 2014, 8, 452–459.
- Neumann, B.; Held, M.; Liebel, U.; Erfle, H.; Rogers, P.; Pepperkok, R.; Ellenberg, J. High-throughput RNAi screening by time-lapse imaging of live human cells. Nat. Methods 2006, 3, 385–390.
- Liu, Y.; Fares, M.; Dunham, N.P.; Gao, Z.; Miao, K.; Jiang, X.; Bollinger, S.S.; Boal, A.K.; Zhang, X. AgHalo: A Facile Fluorogenic Sensor to Detect Drug-Induced Proteome Stress. Angew. Chem. Int. Ed. 2017, 56, 8672–8676.
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44.
- Chakravorty, S.J.; Chan, J.; Greenwood, M.N.; Popa-Burke, I.; Remlinger, K.S.; Pickett, S.D.; Green, D.V.S.; Fillmore, M.C.; Dean, T.W.; Luengo, J.I.; et al. Nuisance Compounds, PAINS Filters, and Dark Chemical Matter in the GSK HTS Collection. SLAS Discov. Adv. Sci. Drug Discov. 2018, 23, 532–545.
- Jasial, S.; Hu, Y.; Bajorath, J. How Frequently Are Pan-Assay Interference Compounds Active? Large-Scale Analysis of Screening Data Reveals Diverse Activity Profiles, Low Global Hit Frequency, and Many Consistently Inactive Compounds. J. Med. Chem. 2017, 60, 3879–3886.
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740.
- Gul, N.; Karlsson, J.; Tängemo, C.; Linsefors, S.; Tuyizere, S.; Perkins, R.; Ala, C.; Zou, Z.; Larsson, E.; Bergö, M.O.; et al. The MTH1 inhibitor TH588 is a microtubule-modulating agent that eliminates cancer cells by activating the mitotic surveillance pathway. Sci. Rep. 2019, 9, 14667.
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 2016, 13, 521–527.