DispHred is the first disorder predictor that includes the effect of the solution pH in its calculations. The DispHred key feature is profiling the pH-dependence disorder of a protein sequence across a pH interval, identifying pH-induced order/disorder protein transitions. DispHred is freely available for academic users at https://ppmclab.pythonanywhere.com/DispHred.
- intrinsically disordered proteins
- pH
- bioinformatics
- disorder prediction
- conditional folding
- machine learning
Conceptual framework
Intrinsically disordered proteins (IDPs) are a class of polypeptides that do not require a defined folded structure to execute their biological activities [1][2][3]. IDPs' unfolded nature is intrinsically encoded in their primary sequence, which is generally enriched in ionizable and polar residues and depleted of hydrophobic amino acids [4]. Thus, IDPs' extended conformation depends both on electrostatic repulsions between uncompensated charges and on a low hydrophobicity, which prevents extensive protein compaction [5]. Based on the balance between attractive and repulsive forces in IDPs, Uversky and coworkers proposed that IDPs populated a distinct region in the mean net charge–hydropathy (C–H) phase space diagram and demonstrated that an empirical boundary line could discriminate folded and disordered proteins [5]. Under that premise, a polypeptide sequence's disordered nature can be predicted by evaluating its C–H relationship in the aforementioned attraction-repulsion scheme. The C–H plot analysis has been extensively applied for disorder prediction.
However, the C–H relationship of a given protein can be modulated by extrinsic factors to the sequence, inducing order/disorder protein transitions. In Santos et al. 2020 [6], we revisited the C–H concept, on the evidence that both protein net charge and hydrophobicity are dependent on pH. We used a machine learning strategy to delineate the most discriminative boundary condition, which was highly similar to the one previously described by Uversky [5]. Therefore, we demonstrated that IDPs' pH-induced folding could be predicted by evaluating the C–H space diagram's pH dependence.
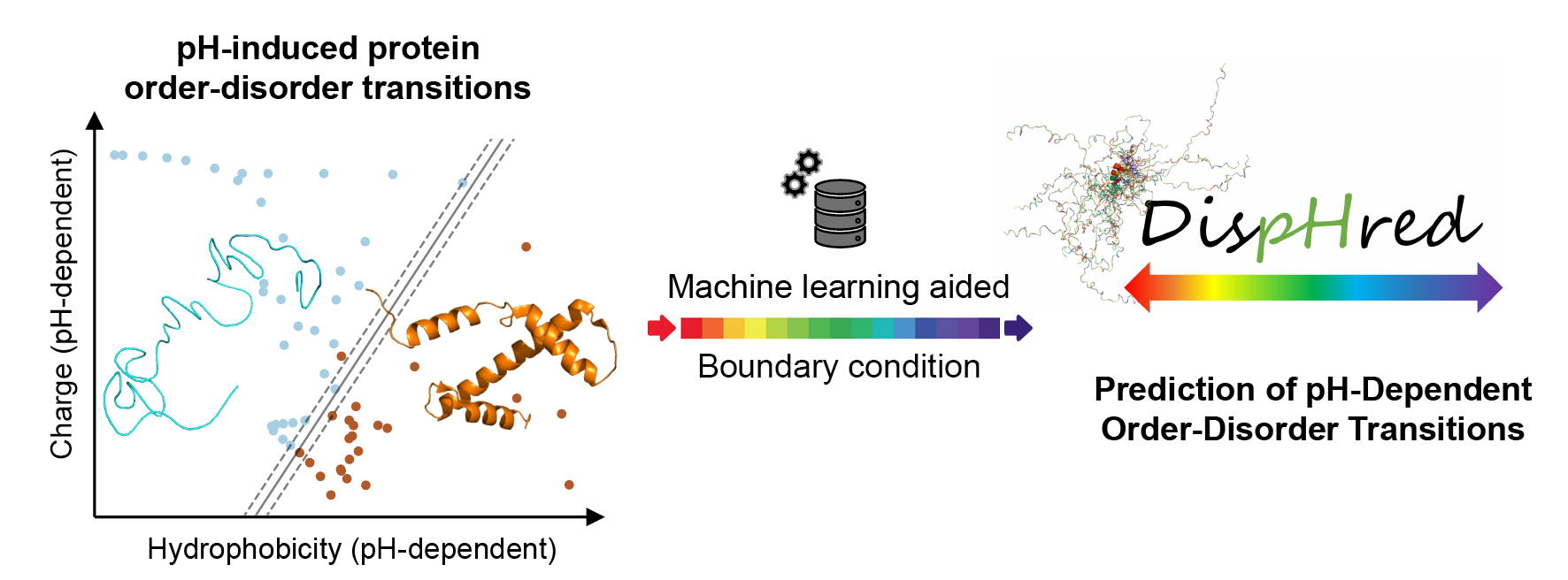
Predictor
DispHred was developed on the basis of the analysis mentioned above and is the first computational tool for predicting protein disorder to consider the solution pH implicitly. DispHred computes the protein net charge per residue and the mean hydrophobicity of a sequence as a function of pH. Then, DispHred applies a boundary condition to separate folded and disordered proteins. DispHred positive values correspond to sequences classified as folded and negative values to those predicted as disordered at the analyzed pH or pH range.
Future perspectives
DispHred is the first disorder predictor dedicated to evaluating the solution's effect and constitutes a proof-of-concept for the implementation of this kind of approach in future prediction methods. Intrinsically disorder tags are increasingly used to solubilize proteins and to tweak the pharmacological properties of protein and peptide pharmaceuticals [7]. We envision that DispHred can be of significant help in these kinds of biotechnological applications.
Notably, the analysis of the local or global hydrophobicity of protein sequences is a pivotal stage in many bioinformatics pipelines aimed to predict protein disorder and its associated properties. Sequence hydrophobicity is strongly modulated by pH, but this effect has been mostly neglected. Our results indicate that by applying a recently developed pH-dependent hydropathy scale, this physicochemical property's contribution to disorder prediction can be extended to the full pH scale. Thus, the implementation of pH-dependent hydropathy scales, like the one we use here, may portray more real-life scenarios to current state-of-the-art disorder predicting algorithms.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21165814
References
- A.Keith Dunker; Zoran Obradovic; The protein trinity—linking function and disorder. Nature Biotechnology 2001, 19, 805-806, 10.1038/nbt0901-805.
- Vivek Kulkarni; Prakash Kulkarni; Intrinsically disordered proteins and phenotypic switching: Implications in cancer.. Progress in Molecular Biology and Translational Science 2019, 166, 63-84, 10.1016/bs.pmbts.2019.03.013.
- Jianhan Chen; Richard W. Kriwacki; Intrinsically Disordered Proteins: Structure, Function and Therapeutics. Journal of Molecular Biology 2018, 430, 2275-2277, 10.1016/j.jmb.2018.06.012.
- H. Jane Dyson; Making Sense of Intrinsically Disordered Proteins. Biophysical Journal 2016, 110, 1013-1016, 10.1016/j.bpj.2016.01.030.
- Vladimir N. Uversky; Joel R. Gillespie; Anthony L. Fink; Why are ?natively unfolded? proteins unstructured under physiologic conditions?. Proteins: Structure, Function, and Bioinformatics 2000, 41, 415-427, 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7.
- Jaime Santos; Valentín Iglesias; Carlos Pintado; Juan Santos-Suárez; Salvador Ventura; DispHred: A Server to Predict pH-Dependent Order–Disorder Transitions in Intrinsically Disordered Proteins. International Journal of Molecular Sciences 2020, 21, 5814, 10.3390/ijms21165814.
- David Paul Minde; Els F Halff; Sander Tans; Designing disorder. Intrinsically Disordered Proteins 2013, 1, e26790, 10.4161/idp.26790.