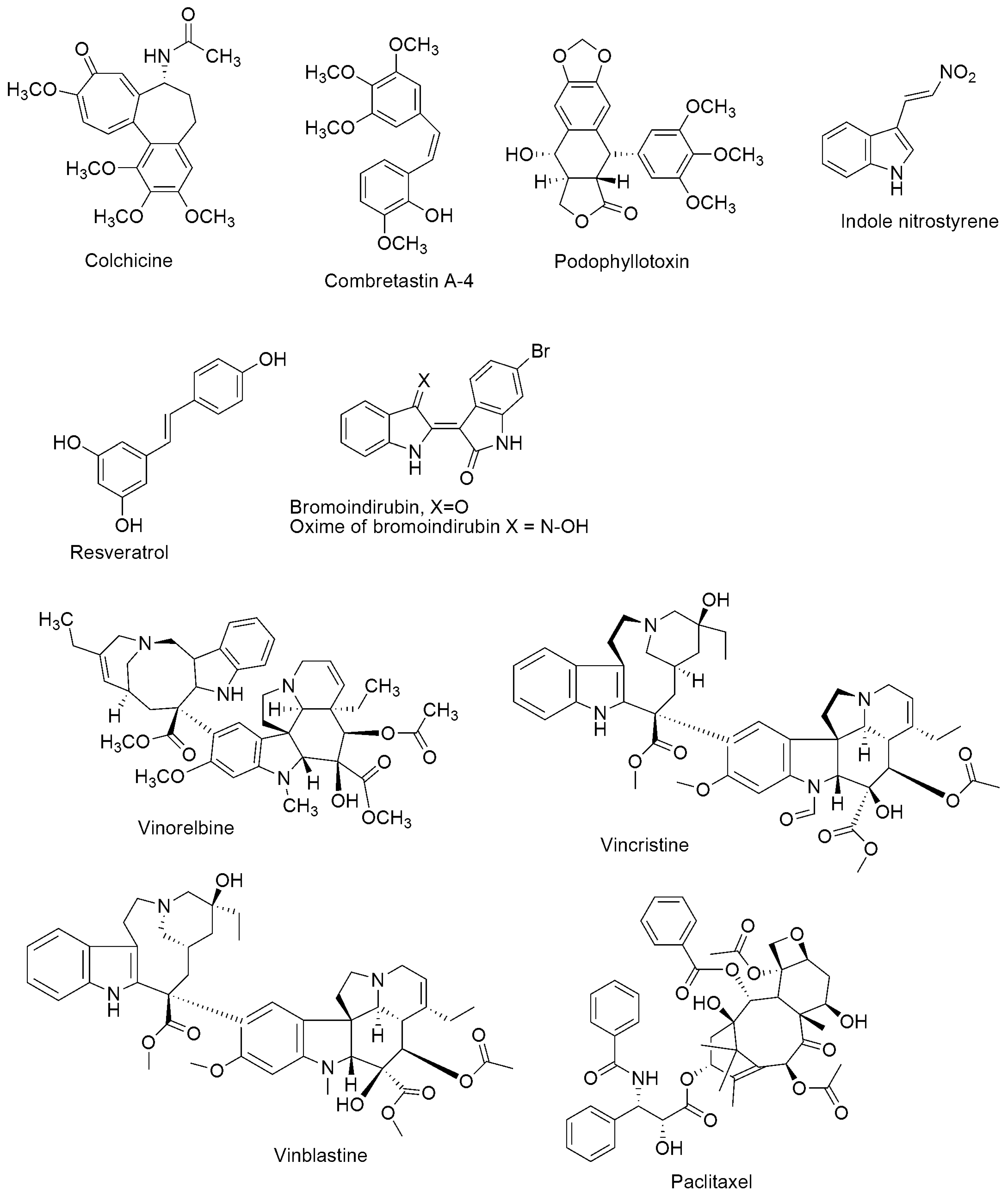
Microtubules are cylindrical protein polymers formed from αβ-tubulin heterodimers in the cytoplasm of eukaryotic cells. Microtubule disturbance may cause cell cycle arrest in the G2/M phase, and anomalous mitotic spindles will form. Microtubules are an important target for cancer drug action because of their critical role in mitosis. Several microtubule-targeting agents with vast therapeutic advantages have been developed, but they often lead to multidrug resistance and adverse side effects. Thus, single-target therapy has drawbacks in the effective control of tubulin polymerization. Molecular hybridization, based on the amalgamation of two or more pharmacophores of bioactive conjugates to engender a single molecular structure with enhanced pharmacokinetics and biological activity, compared to their parent molecules, has recently become a promising approach in drug development. The practical application of combined active scaffolds targeting tubulin polymerization inhibitors has been corroborated in the past few years
- tubulin
- cancer
- microtubules
1. Introduction
Microtubules are cylindrical protein polymers assembled in the cytoplasm of all eukaryotic cells by polymerization of αβ-tubulin heterodimers. Microtubules are characterized by a unique dynamic instability process, which involves continuously alternating phases of elongation and shortening interspersed with catastrophic disassembly events. In living cells, the minus ends of microtubules are associated with structures called microtubule organizing centers (MTOCs) [1]. The main MTOC in the cell is the centrosome, contiguously positioned to the nucleus. Microtubules play an essential function in the maintenance of cell shape and in the progression of cell division [2]. They are the principal components for the formation of the mitotic spindles. They all participate in signaling, intracellular transport, and cell motility [3]. Disturbance of microtubules may induce cell cycle arrest at the G2/M phase. Their pre-eminent importance in force generation in mitosis to enable the segregation of chromosomes makes microtubules attractive as intra-cellular targets for anti-cancer drug action.
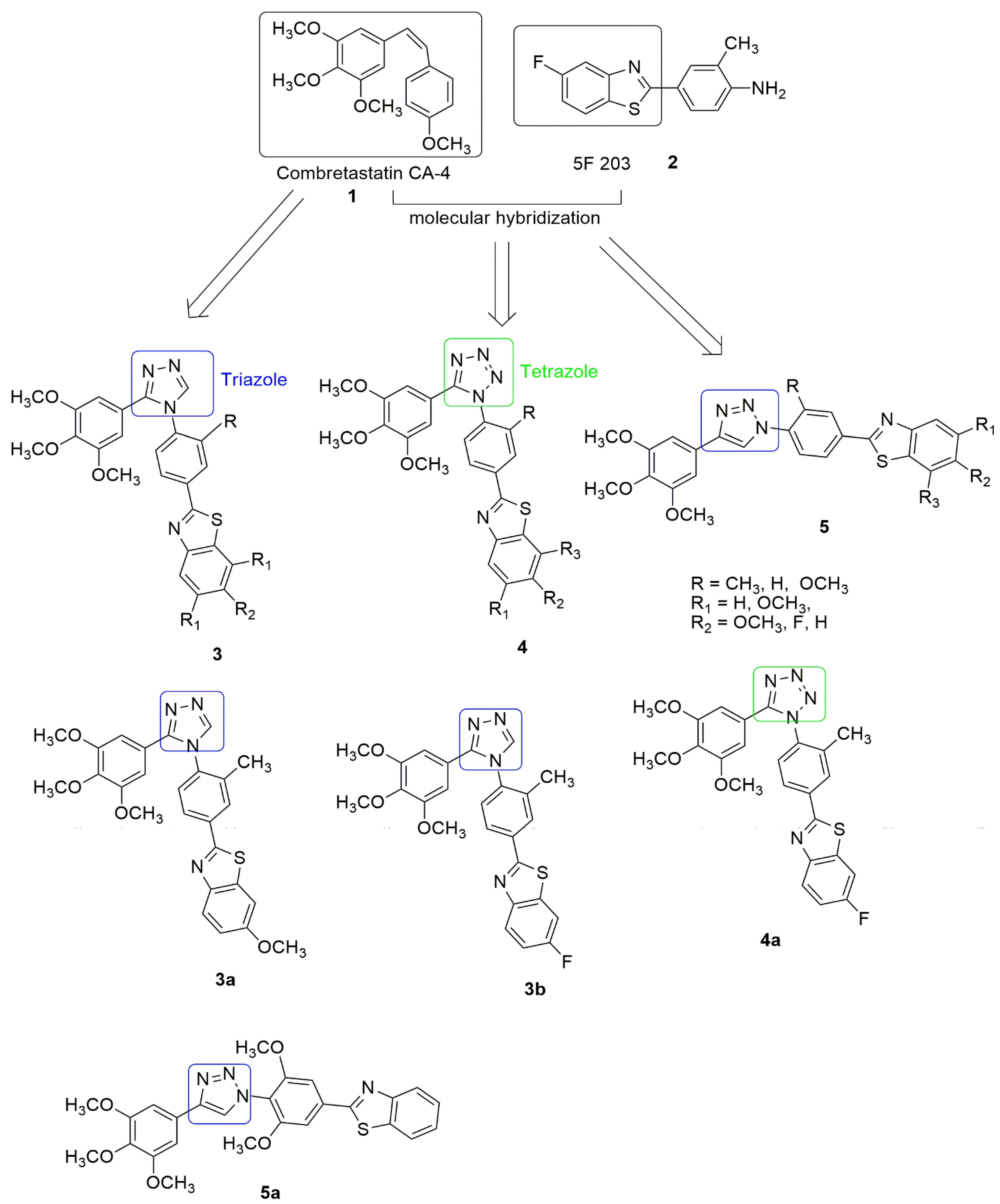
2. Tubulin Polymerization Hybrids
Triazole and Tetrazole Hybrids
3. Benzimidazole Hybrids
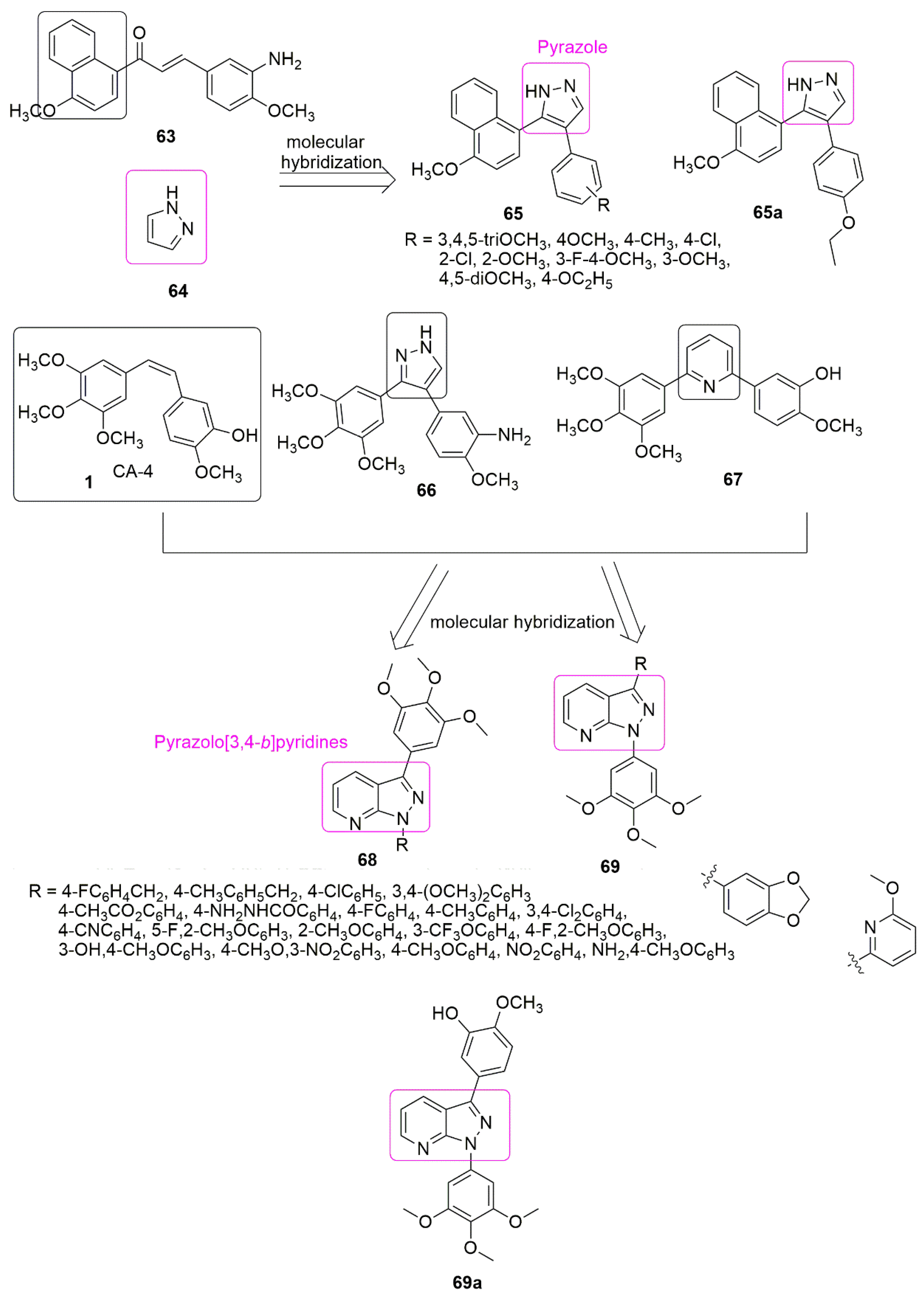
4. Pyrazole Hybrids
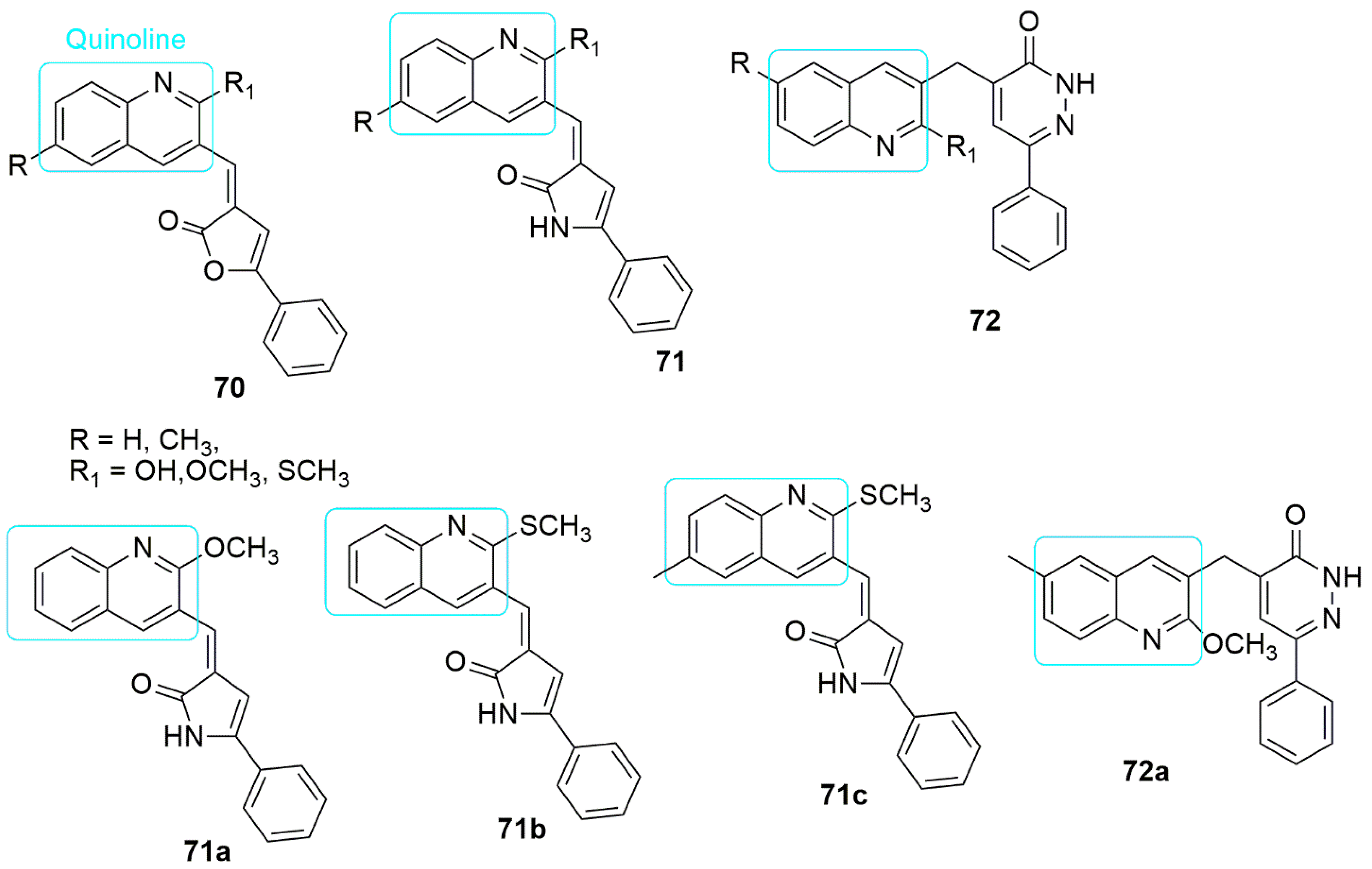
5. Quinoline Hybrids
6. Quinazolinone Hybrids
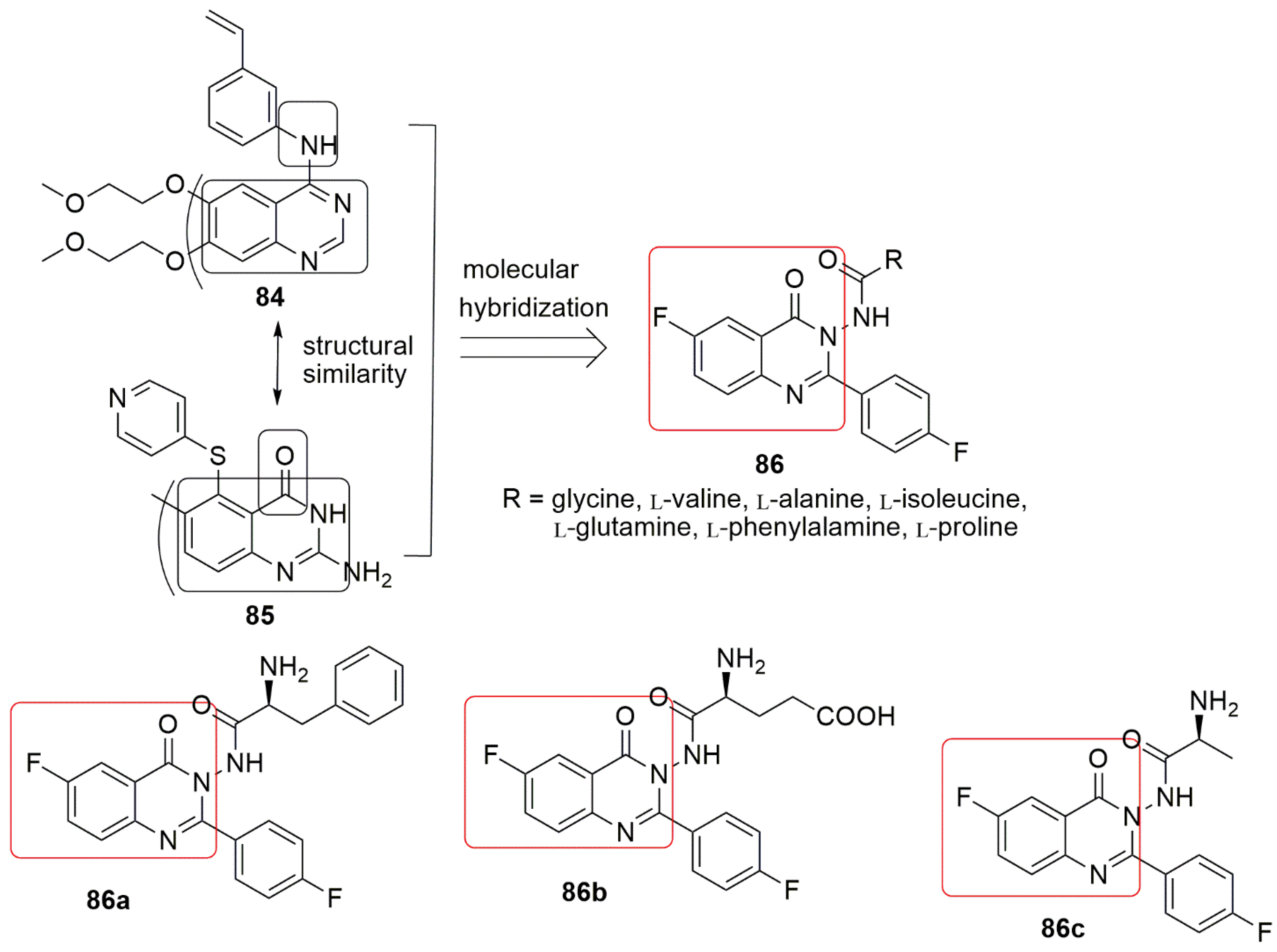
7. Quinolinone Hybrids
8. Chalcone Hybrids
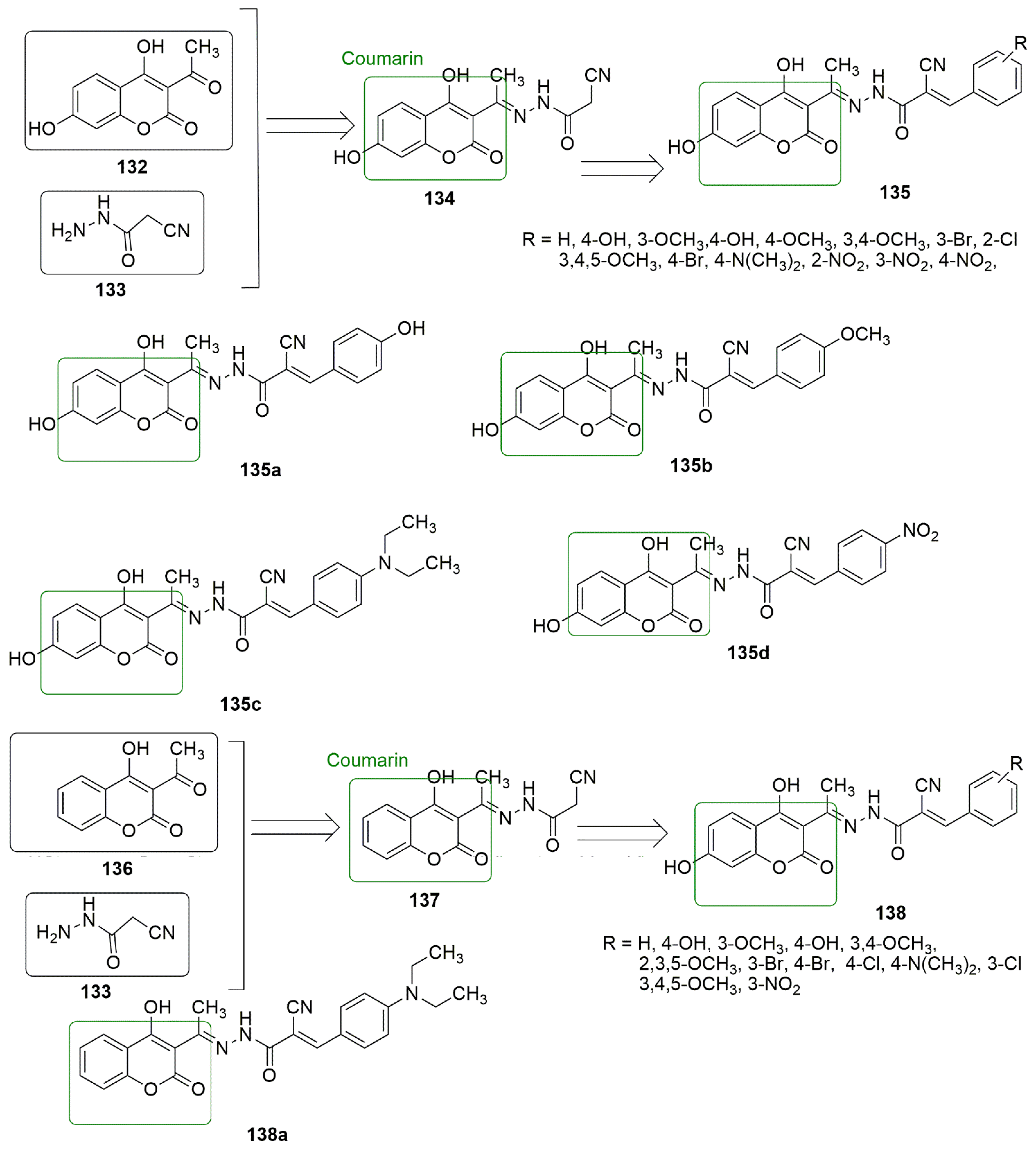
9. Coumarin Hybrids
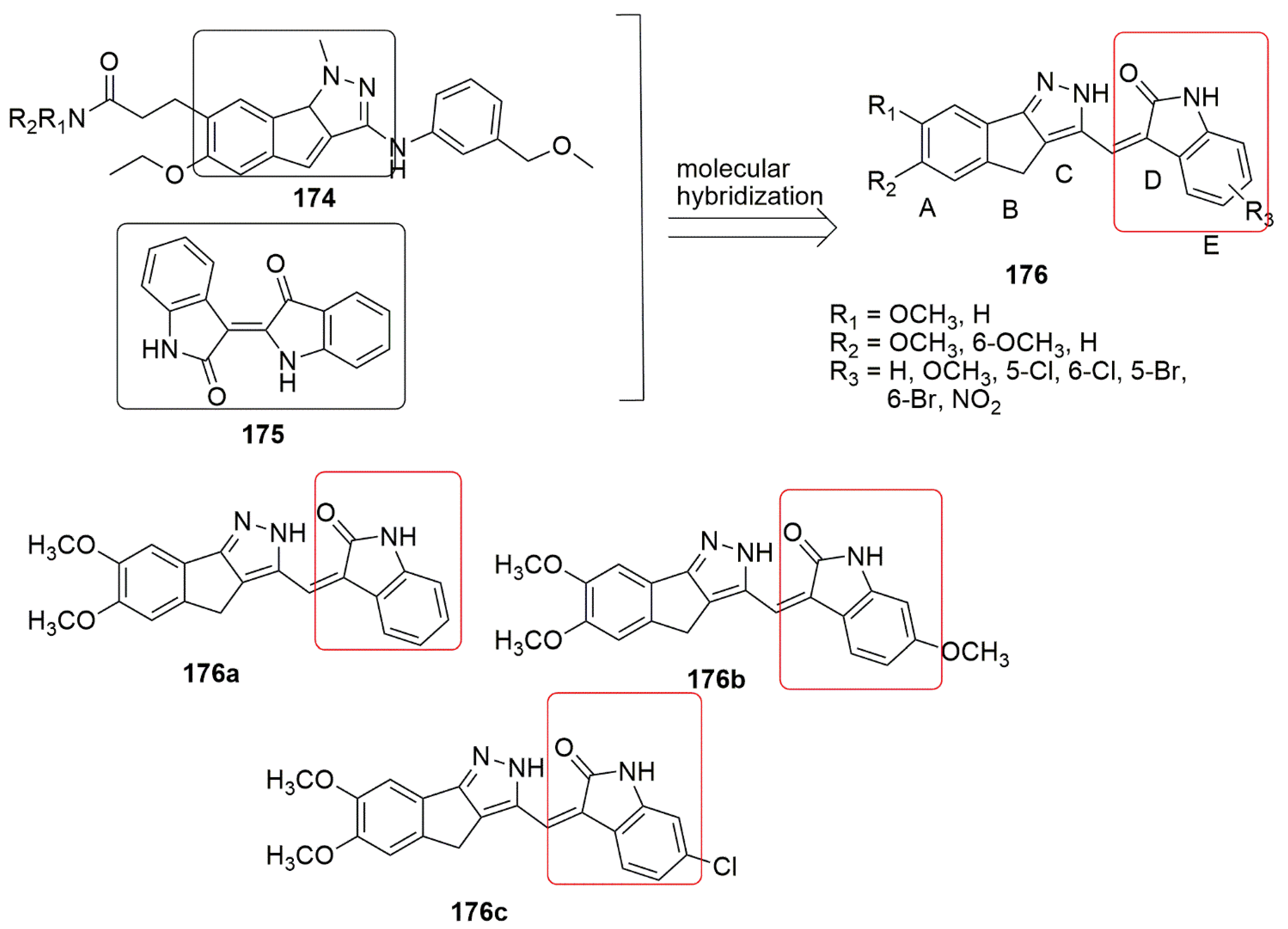
10. Indole Hybrids
11. Oxindole Hybrids
12. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/ijms23074001