KMT2A (Lysine methyltransferase 2A) is a member of the epigenetic machinery, encoding a lysine methyltransferase responsible for the transcriptional activation through lysine 4 of histone 3 (H3K4) methylation. KMT2A has a crucial role in gene expression, thus it is associated to pathological conditions when found mutated. KMT2A germinal mutations are associated to Wiedemann–Steiner syndrome and also in patients with initial clinical diagnosis of several other chromatinopathies (i.e., Coffin–Siris syndromes, Kabuki syndrome, Cornelia De Lange syndrome, Rubinstein–Taybi syndrome), sharing an overlapping phenotype. On the other hand, KMT2A somatic mutations have been reported in several tumors, mainly blood malignancies. Due to its evolutionary conservation, the role of KMT2A in embryonic development, hematopoiesis and neurodevelopment has been explored in different animal models, and epigenetic treatments for disorders linked to KMT2A dysfunction have been extensively investigated.
- KMT2A
- chromatinopathies
- tumors
- Wiedemann–Steiner syndrome
1. Introduction
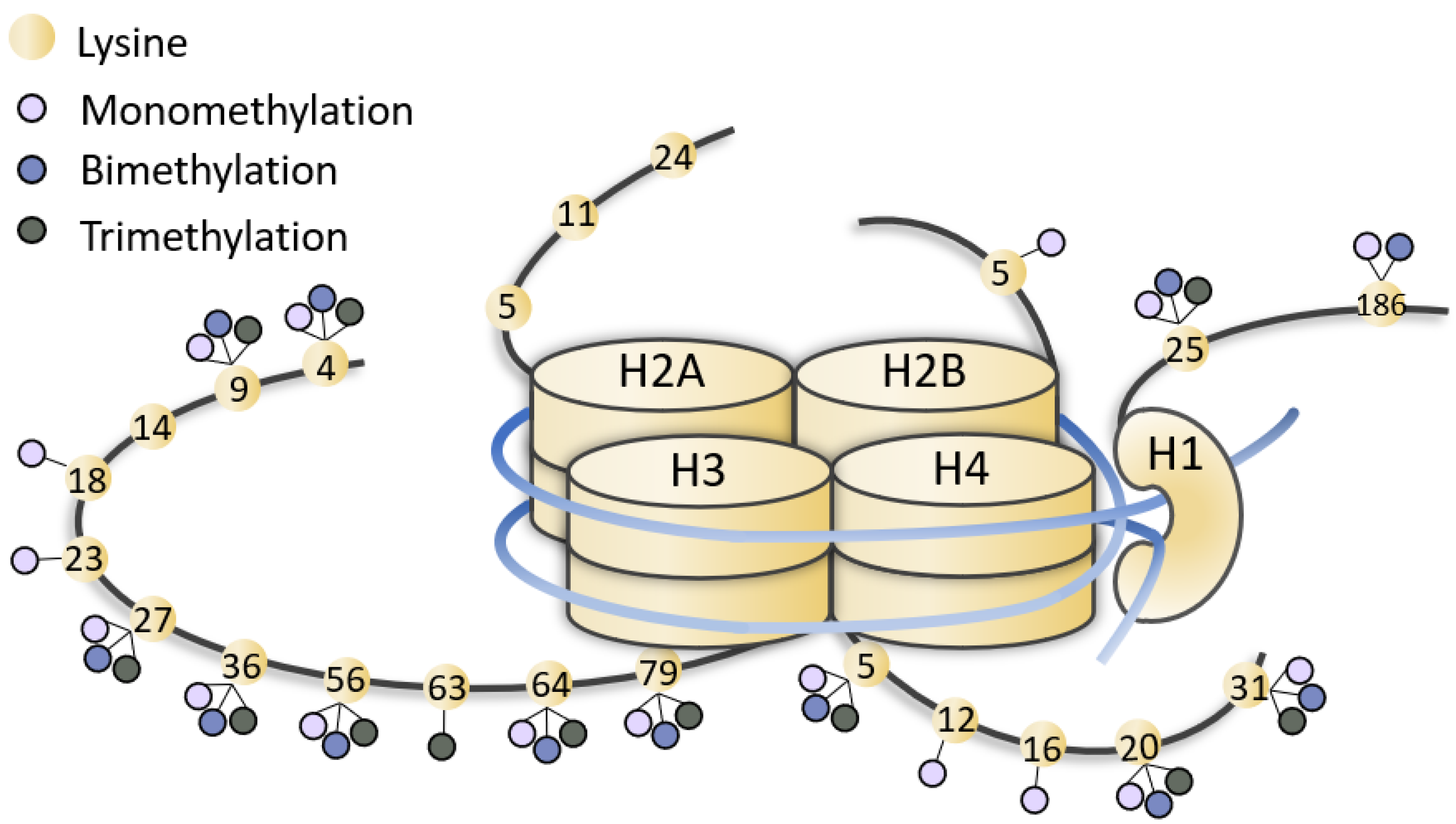
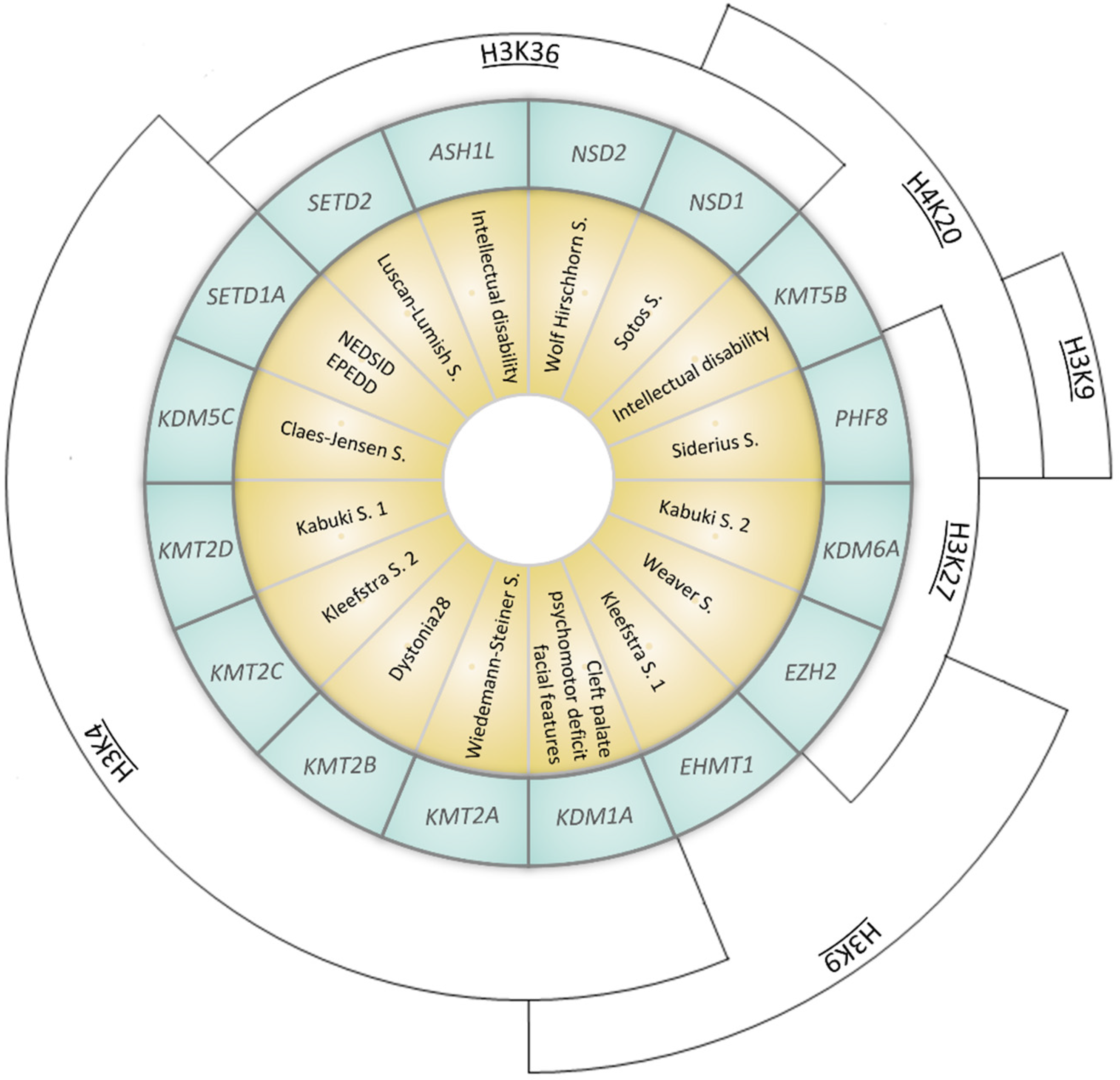
| Gene (OMIM *) | Associated Developmental Disorder(s) (OMIM #) | Targeted Lysine Residue |
|---|---|---|
| SETD1A (611052) | Neurodevelopmental disorder with speech impairment and dysmorphic facies NEDSID (619056)/Epilepsy, early-onset, with or without developmental delay EPEDD (618832) | H3K4 (met) |
| SETD2 (612778) | Luscan-Lumish S. (616831) | H3K36 (met) |
| KDM1A (609132) | Cleft palate, psychomotor retardation, distinctive facial features (616728) | H3K4 (demet) H3K9 (demet) |
| KDM5C (314690) | Claes-Jensen S. (300534) | H3K4 (demet) |
| KDM6A (300128) | Kabuki S. 2 (300867) | H3K27 (demet) |
| KMT2A (159555) | Wiedemann–Steiner S. (605130) | H3K4 (met) |
| KMT2B (606834) | Dystonia 28 (617284) | H3K4 (met) |
| KMT2C (606833) | Kleefstra S. 2 (617768) | H3K4 (met) |
| KMT2D (602113) | Kabuki S. 1 (147920) | H3K4 (met) |
| KMT5B (610881) | Intellectual disability (617788) | H4K20 (met) |
| EZH2 (601573) | Weaver S. (277590) | H3K9 (met) H3K27 (met) |
| EHMT1 (607001) | Kleefstra S. 1 (610253) | H3K9 (met) |
| ASH1L (607999) | Intellectual disability (617796) | H3K36 (met) |
| NSD1 (606681) | Sotos S. (117550) | H3K36 (met) H4K20 (met) |
| NSD2 (602952) | Wolf Hirschhorn S. (194190) | H3K36 (met) |
| PHF8 (300560) | Siderius S. (300263) | H3K9 (demet) H3K27 (demet) H4K20 (demet) |
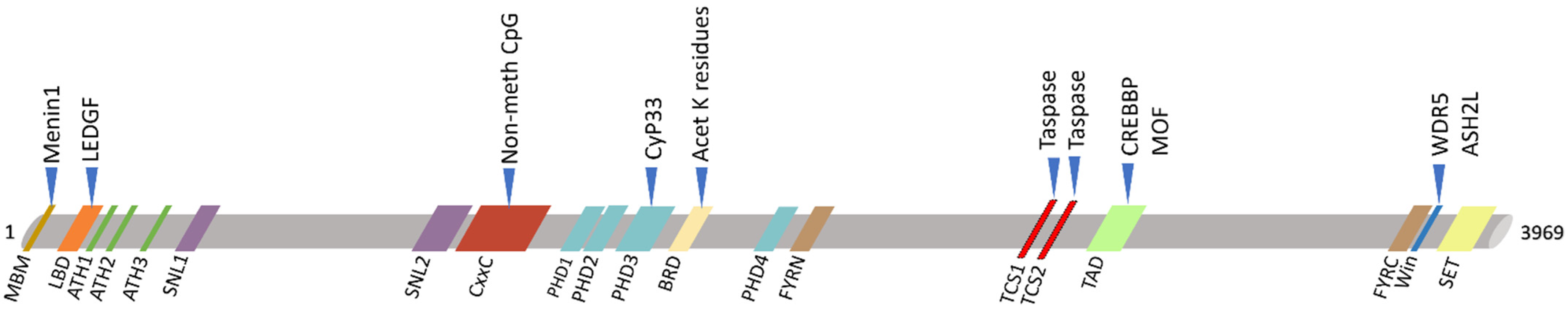
2. KMT2A Germline Mutations
2.1. Wiedemann–Steiner Syndrome
| WDSTS | CdLS | CSS | KS | RSTS | |
|---|---|---|---|---|---|
| [26] | 1 + 1 pt [27][28] | 1 pt [29] | 2 pt [30] | 1 + 6 pt [26][31] | |
| Vision problems | − | 0/2 | 1/1 | 1/2 | 1/7 |
| Cardiac problems | + | 1/2 | 1/1 | 1/2 | 0/7 |
| CNS problems | +/− | 1/2 | 0/1 | NA | 0/7 |
| Genitourinary problems | − | 0/2 | 1/1 | 1/2 | 2/7 |
| Feeding problems | + | 0/2 | 1/1 | 1/2 | 3/7 |
| Behavior problems | + | 1/2 | 0/1 | NA | 3/7 |
| Frequent infection | − | 0/2 | 1/1 | 1/2 | 0/7 |
| Seizures | +/− | 0/2 | 0/1 | 1/2 | 1/7 |
| ID | ++ | 2/2 | 1/1 | 1/2 | 7/7 |
| Speech delay | ++ | 1/2 | 1/1 | NA | 5/7 |
| Microcephaly | − | 2/2 | NA | NA | 3/7 |
| Eyes anomalies (thick eyebrows, synophrys, long eyelashes, ptosis, downslanting/narrow palpebral fissure) | + | 2/2 | 1/1 | 2/2 | 7/7 |
| Nose anomalies (depressed nasal bridge, broad nasal tip) | + | 2/2 | 1/1 | 2/2 | 7/7 |
| Mouth anomalies (high arched palate, thin upper vermilion) | +/− | 2/2 | 1/1 | 0/2 | 4/7 |
| Hands/feet anomalies (clinodactyly, brachydactyly, persistent fetal finger pads, broad halluces) | +/− | 2/2 | 1/1 | 2/2 | 6/7 |
| Delayed bone age | + | 0/2 | NA | NA | 0/7 |
| Hirsutism | + | 1/2 | 1/1 | NA | 4/7 |
| Hypotonia | ++ | NA | 1/1 | 2/2 | 3/7 |
2.2. Other Chromatinopathies
Mutations in KMT2A have been also found in patients with a clinical presentation suggestive of other chromatinopathies (i.e., Coffin–Siris syndromes, Kabuki syndrome, Cornelia De Lange syndrome, Rubinstein–Taybi syndrome) but negative for alterations in the related known-causative genes. Their clinical presentation shares with WDSTS some phenotypic features and it is caused by alterations of genes involved in the regulation and maintenance of chromatin state as KMT2A. Indeed, these syndromes are caused by mutations in genes of the epigenetic machinery and therefore are known as chromatinopathies [16][18].
3. KMT2A Somatic Mutations
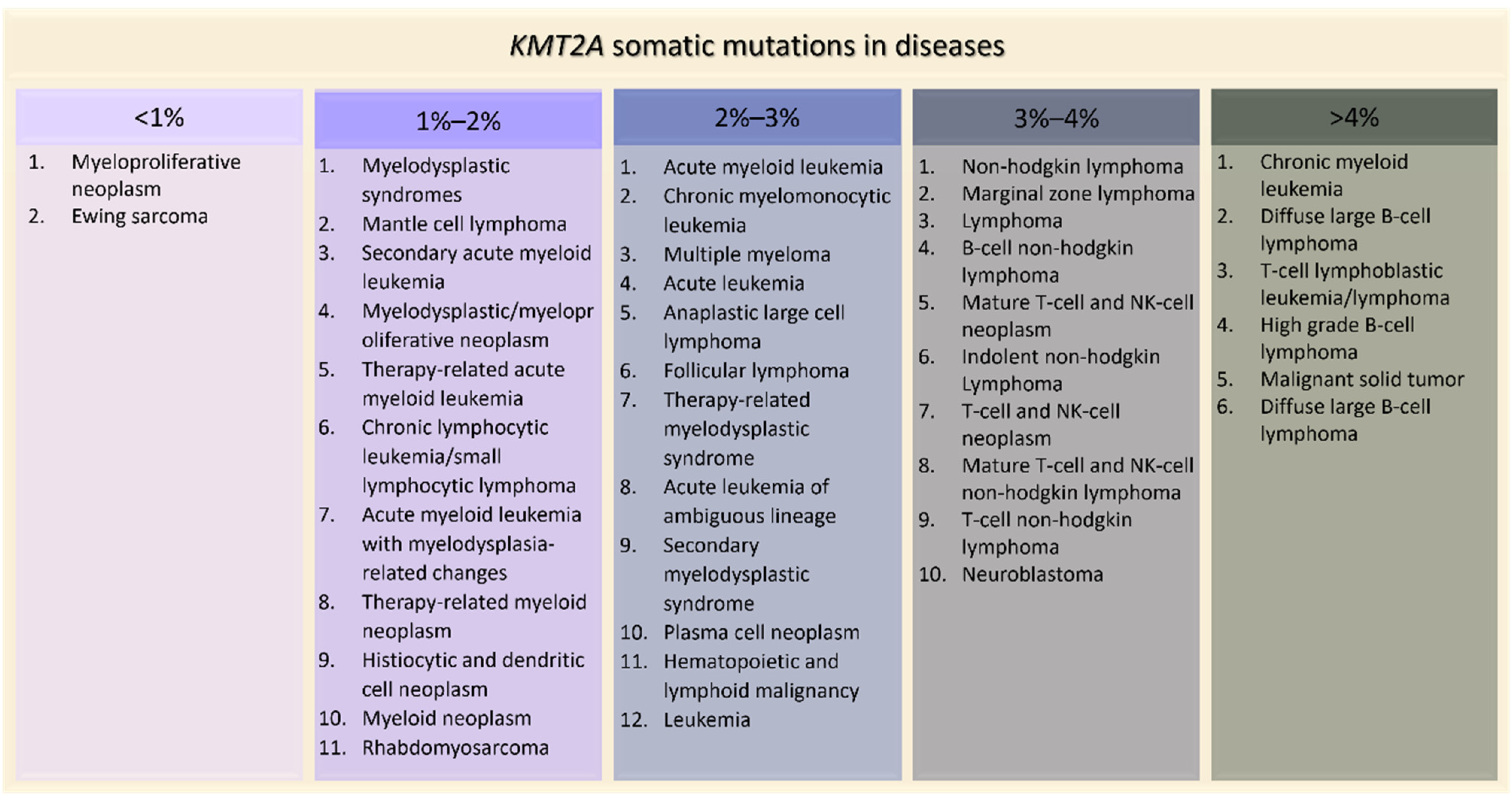 Figure 5. KMT2A somatic mutations in tumors ordered by percentage of positive cases (AACR Project GENIE).
Figure 5. KMT2A somatic mutations in tumors ordered by percentage of positive cases (AACR Project GENIE).4. Effects of KMT2A Mutations in Animal Models
5. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/genes13030514
References
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705.
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell 2012, 48, 491–507.
- Garcia, B.A.; Hake, S.B.; Diaz, R.L.; Kauer, M.; Morris, S.A.; Recht, J.; Shabanowitz, J.; Mishra, N.; Strahl, B.D.; Allis, C.D.; et al. Organismal Differences in Post-Translational Modifications in Histones H3 and H4. J. Biol. Chem. 2007, 282, 7641–7655.
- Trojer, P.; Zhang, J.; Yonezawa, M.; Schmidt, A.; Zheng, H.; Jenuwein, T.; Reinberg, D. Dynamic Histone H1 Isotype 4 Methylation and Demethylation by Histone Lysine Methyltransferase G9a/KMT1C and the Jumonji Domain-Containing JMJD2/KDM4 Proteins. J. Biol. Chem. 2009, 284, 8395–8405.
- Daujat, S.; Weiss, T.; Mohn, F.; Lange, U.C.; Ziegler-Birling, C.; Zeissler, U.; Lappe, M.; Schübeler, D.; Torres-Padilla, M.E.; Schneider, R. H3K64 Trimethylation Marks Heterochromatin and Is Dynamically Remodeled during Developmental Reprogramming. Nat. Struct. Mol. Biol. 2009, 16, 777–781.
- Weiss, T.; Hergeth, S.; Zeissler, U.; Izzo, A.; Tropberger, P.; Zee, B.M.; Dundr, M.; Garcia, B.A.; Daujat, S.; Schneider, R. Histone H1 Variant-Specific Lysine Methylation by G9a/KMT1C and Glp1/KMT1D. Epigenet. Chromatin 2010, 3, 7.
- Rodríguez-Paredes, M.; Esteller, M. Cancer Epigenetics Reaches Mainstream Oncology. Nat. Med. 2011, 17, 330–339.
- van Aller, G.S.; Reynoird, N.; Barbash, O.; Huddleston, M.; Liu, S.; Zmoos, A.F.; McDevitt, P.; Sinnamon, R.; Le, B.C.; Mas, G.; et al. Smyd3 Regulates Cancer Cell Phenotypes and Catalyzes Histone H4 Lysine 5 Methylation. Epigenetics 2012, 7, 340–343.
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med. 2017, 49, e324.
- Husmann, D.; Gozani, O. Histone Lysine Methyltransferases in Biology and Disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889.
- Liu, Y.; Qin, S.; Chen, T.Y.; Lei, M.; Dhar, S.S.; Ho, J.C.; Dong, A.; Loppnau, P.; Li, Y.; Lee, M.G.; et al. Structural Insights into Trans-Histone Regulation of H3K4 Methylation by Unique Histone H4 Binding of MLL3/4. Nat. Commun. 2019, 10, 36.
- Guo, L.; Lee, Y.T.; Zhou, Y.; Huang, Y. Targeting Epigenetic Regulatory Machinery to Overcome Cancer Therapy Resistance. Semin. Cancer Biol. 2021, in press.
- Zhang, T.; Zhang, W.; Liu, L.; Chen, Y. Simultaneous Detection of Site-Specific Histone Methylations and Acetylation Assisted by Single Template Oriented Molecularly Imprinted Polymers. Analyst 2020, 145, 1376–1383.
- Di Nisio, E.; Lupo, G.; Licursi, V.; Negri, R. The Role of Histone Lysine Methylation in the Response of Mammalian Cells to Ionizing Radiation. Front. Genet. 2021, 12, 482.
- Khare, S.P.; Habib, F.; Sharma, R.; Gadewal, N.; Gupta, S.; Galande, S. HIstome—A Relational Knowledgebase of Human Histone Proteins and Histone Modifying Enzymes. Nucleic Acids Res. 2012, 40, D337–D342.
- Fahrner, J.A.; Bjornsson, H.T. Mendelian Disorders of the Epigenetic Machinery: Tipping the Balance of Chromatin States. Annu. Rev. Genom. Hum. Genet. 2014, 15, 269–293.
- Bjornsson, H.T. The Mendelian Disorders of the Epigenetic Machinery. Genome Res. 2015, 25, 1473–1481.
- Fahrner, J.A.; Bjornsson, H.T. Mendelian Disorders of the Epigenetic Machinery: Postnatal Malleability and Therapeutic Prospects. Hum. Mol. Genet. 2019, 28, R254–R264.
- Faundes, V.; Newman, W.G.; Bernardini, L.; Canham, N.; Clayton-Smith, J.; Dallapiccola, B.; Davies, S.J.; Demos, M.K.; Goldman, A.; Gill, H.; et al. Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am. J. Hum. Genet. 2018, 102, 175–187.
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol. Cell. Proteom. MCP 2014, 13, 397–406.
- Hess, J.L. MLL: A Histone Methyltransferase Disrupted in Leukemia. Trends Mol. Med. 2004, 10, 500–507.
- Southall, S.M.; Wong, P.S.; Odho, Z.; Roe, S.M.; Wilson, J.R. Structural Basis for the Requirement of Additional Factors for MLL1 SET Domain Activity and Recognition of Epigenetic Marks. Mol. Cell 2009, 33, 181–191.
- Wiedemann, H.R.; Kunze, J.; Grosse, F.R.; Dibbern, H. A Syndrome of Abnormal Facies, Short Stature, and Psychomotor Retardation. In Atlas of Clinical Syndromes: A Visual Aid to Diagnosis for Clinicians and Practicing Physicians, 2nd ed.; Wolfe Publishing Ltd.: London, UK, 1989; pp. 198–199.
- Miyake, N.; Tsurusaki, Y.; Koshimizu, E.; Okamoto, N.; Kosho, T.; Brown, N.J.; Tan, T.Y.; Yap, P.J.J.; Suzumura, H.; Tanaka, T.; et al. Delineation of Clinical Features in Wiedemann-Steiner Syndrome Caused by KMT2A Mutations. Clin. Genet. 2016, 89, 115–119.
- Aggarwal, A.; Rodriguez-Buritica, D.F.; Northrup, H. Wiedemann-Steiner Syndrome: Novel Pathogenic Variant and Review of Literature. Eur. J. Med. Genet. 2017, 60, 285–288.
- di Fede, E.; Massa, V.; Augello, B.; Squeo, G.; Scarano, E.; Perri, A.M.; Fischetto, R.; Causio, F.A.; Zampino, G.; Piccione, M.; et al. Expanding the Phenotype Associated to KMT2A Variants: Overlapping Clinical Signs between Wiedemann–Steiner and Rubinstein–Taybi Syndromes. Eur. J. Hum. Genet. 2021, 29, 88–98.
- Yuan, B.; Pehlivan, D.; Karaca, E.; Patel, N.; Charng, W.-L.; Gambin, T.; Gonzaga-Jauregui, C.; Sutton, V.R.; Yesil, G.; Bozdogan, S.T.; et al. Global Transcriptional Disturbances Underlie Cornelia de Lange Syndrome and Related Phenotypes. J. Clin. Investig. 2015, 125, 636–651.
- Parenti, I.; Teresa-Rodrigo, M.E.; Pozojevic, J.; Ruiz Gil, S.; Bader, I.; Braunholz, D.; Bramswig, N.C.; Gervasini, C.; Larizza, L.; Pfeiffer, L.; et al. Mutations in Chromatin Regulators Functionally Link Cornelia de Lange Syndrome and Clinically Overlapping Phenotypes. Hum. Genet. 2017, 136, 307–320.
- Bramswig, N.C.; Lüdecke, H.J.; Alanay, Y.; Albrecht, B.; Barthelmie, A.; Boduroglu, K.; Braunholz, D.; Caliebe, A.; Chrzanowska, K.H.; Czeschik, J.C.; et al. Exome Sequencing Unravels Unexpected Differential Diagnoses in Individuals with the Tentative Diagnosis of Coffin–Siris and Nicolaides–Baraitser Syndromes. Hum. Genet. 2015, 134, 553–568.
- Sobreira, N.; Brucato, M.; Zhang, L.; Ladd-Acosta, C.; Ongaco, C.; Romm, J.; Doheny, K.F.; Mingroni-Netto, R.C.; Bertola, D.; Kim, C.A.; et al. Patients with a Kabuki Syndrome Phenotype Demonstrate DNA Methylation Abnormalities. Eur. J. Hum. Genet. 2017, 25, 1335–1344.
- Negri, G.; Magini, P.; Milani, D.; Crippa, M.; Biamino, E.; Piccione, M.; Sotgiu, S.; Perrìa, C.; Vitiello, G.; Frontali, M.; et al. Exploring by Whole Exome Sequencing Patients with Initial Diagnosis of Rubinstein–Taybi Syndrome: The Interconnections of Epigenetic Machinery Disorders. Hum. Genet. 2019, 138, 257–269.
- Meyer, C.; Kowarz, E.; Hofmann, J.; Renneville, A.; Zuna, J.; Trka, J.; Ben Abdelali, R.; Macintyre, E.; De Braekeleer, E.; De Braekeleer, M.; et al. New Insights to the MLL Recombinome of Acute Leukemias. Leukemia 2009, 23, 1490–1499.
- Sweeney, S.M.; Cerami, E.; Baras, A.; Pugh, T.J.; Schultz, N.; Stricker, T.; Lindsay, J.; Del Vecchio Fitz, C.; Kumari, P.; Micheel, C.; et al. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831.
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic Cancer Genetics at High-Resolution. Nucleic Acids Res. 2017, 45, D777–D783.
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059.
- Kishtagari, A.; Levine, R.L. The Role of Somatic Mutations in Acute Myeloid Leukemia Pathogenesis. Cold Spring Harb. Perspect. Med. 2021, 11, a034975.
- Mullighan, C.G. Genome Sequencing of Lymphoid Malignancies. Blood 2013, 122, 3899–3907.
- Huang, Y.H.; Cai, K.; Xu, P.P.; Wang, L.; Huang, C.X.; Fang, Y.; Cheng, S.; Sun, X.J.; Liu, F.; Huang, J.Y.; et al. CREBBP/EP300 Mutations Promoted Tumor Progression in Diffuse Large B-Cell Lymphoma through Altering Tumor-Associated Macrophage Polarization via FBXW7-NOTCH-CCL2/CSF1 Axis. Signal Transduct. Target. Ther. 2021, 6, 10.
- Yu, B.D.; Hess, J.L.; Horning, S.E.; Brown, G.A.J.; Korsmeyer, S.J. Altered HOX Expression and Segmental Identity in Mll-Mutant Mice. Nature 1995, 378, 505–508.
- Katsani, K.R.; Arredondo, J.J.; Kal, A.J.; Verrijzer, C.P. A Homeotic Mutation in the Trithorax SET Domain Impedes Histone Binding. Genes Dev. 2001, 15, 2197–2202.
- Yamashita, M.; Hirahara, K.; Shinnakasu, R.; Hosokawa, H.; Norikane, S.; Kimura, M.Y.; Hasegawa, A.; Nakayama, T. Crucial Role of MLL for the Maintenance of Memory T Helper Type 2 Cell Responses. Immunity 2006, 24, 611–622.
- McMahon, K.A.; Hiew, S.Y.L.; Hadjur, S.; Veiga-Fernandes, H.; Menzel, U.; Price, A.J.; Kioussis, D.; Williams, O.; Brady, H.J.M. Mll Has a Critical Role in Fetal and Adult Hematopoietic Stem Cell Self-Renewal. Cell Stem Cell 2007, 1, 338–345.
- Wan, X.; Hu, B.; Liu, J.X.; Feng, X.; Xiao, W. Zebrafish Mll Gene Is Essential for Hematopoiesis. J. Biol. Chem. 2011, 286, 33345–33357.
- Huang, Y.C.; Shih, H.Y.; Lin, S.J.; Chiu, C.C.; Ma, T.L.; Yeh, T.H.; Cheng, Y.C. The Epigenetic Factor Kmt2a/Mll1 Regulates Neural Progenitor Proliferation and Neuronal and Glial Differentiation. Dev. Neurobiol. 2015, 75, 452–462.
- Gupta, S.; Kim, S.Y.; Artis, S.; Molfese, D.L.; Schumacher, A.; Sweatt, J.D.; Paylor, R.E.; Lubin, F.D. Histone Methylation Regulates Memory Formation. J. Neurosci. 2010, 30, 3589–3599.
- Kerimoglu, C.; Sakib, M.S.; Jain, G.; Benito, E.; Burkhardt, S.; Capece, V.; Kaurani, L.; Halder, R.; Agís-Balboa, R.C.; Stilling, R.; et al. KMT2A and KMT2B Mediate Memory Function by Affecting Distinct Genomic Regions. Cell Rep. 2017, 20, 538–548.
- Jakovcevski, M.; Ruan, H.; Shen, E.Y.; Dincer, A.; Javidfar, B.; Ma, Q.; Peter, C.J.; Cheung, I.; Mitchell, A.C.; Jiang, Y.; et al. Neuronal Kmt2a/Mll1 Histone Methyltransferase Is Essential for Prefrontal Synaptic Plasticity and Working Memory. J. Neurosci. 2015, 35, 5097–5108.
- Shen, E.Y.; Jiang, Y.; Javidfar, B.; Kassim, B.; Loh, Y.H.E.; Ma, Q.; Mitchell, A.C.; Pothula, V.; Stewart, A.F.; Ernst, P.; et al. Neuronal Deletion of Kmt2a/Mll1 Histone Methyltransferase in Ventral Striatum Is Associated with Defective Spike-Timing-Dependent Striatal Synaptic Plasticity, Altered Response to Dopaminergic Drugs, and Increased Anxiety. Neuropsychopharmacology 2016, 41, 3103–3113.
- Jakovcevski, M.; Akbarian, S. Epigenetic Mechanisms in Neurological Disease. Nat. Med. 2012, 18, 1194–1204.
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting Epigenetic Regulators for Cancer Therapy: Mechanisms and Advances in Clinical Trials. Signal Transduct. Target. Ther. 2019, 4, 62.
- Fagan, R.J.; Dingwall, A.K. COMPASS Ascending: Emerging Clues Regarding the Roles of MLL3/KMT2C and MLL2/KMT2D Proteins in Cancer. Cancer Lett. 2019, 458, 56–65.
- Cheng, L.H.; Liu, Y.W.; Wu, C.C.; Wang, S.; Tsai, Y.C. Psychobiotics in Mental Health, Neurodegenerative and Neurodevelopmental Disorders. J. Food Drug Anal. 2019, 27, 632–648.