Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
In vitro models of traumatic brain injury (TBI) help to elucidate the pathological mechanisms responsible for cell dysfunction and death. Mitochondrial Ca2+ overload and a drop in ΔΨm may cause delayed neuronal death and thus play a key role in the development of the post-traumatic syndrome. Preventing prolonged ΔΨm depolarization may be a promising therapeutic approach to improve neuronal survival after traumatic brain injury.
- mechanical trauma
- post-traumatic syndrome
- neuroglial culture
- β-III tubulin
- GFAP
- [Ca2+]i
- ΔΨm
- NMDAR
1. Introduction
Pathological mechanisms underlying the detrimental effects of traumatic brain injury (TBI) are divided into primary and secondary injury mechanisms. Primary damage is due to the mechanical impact itself (force, degree of penetration, and area of damage) and develops in the first few seconds after the trauma. The most important aspect of this stage is necrotic cell death in the area of the injured brain tissue. Secondary damage is longer-term (hours-days) and is mediated by primary damage mechanisms [1]. They include pathological depolarization due to shear stress, changes in cerebral blood flow (CBF) dynamics, as well as neuro-inflammatory processes [2] caused by glial and immune cells coming from the bloodstream during blood–brain barrier (BBB) permeabilization.
At the same time, the central mechanism of long-term neuronal death induced by TBI is glutamate excitotoxicity [3]. Briefly, the exposure of neural tissue to kinetic energy leads to the immediate depolarization of neurons, causing an abundant release of excitatory neurotransmitters, the most prevalent of which is glutamate [4][5]. It was previously shown that concussion leads to a 50-fold local increase in the concentration of extracellular glutamate in the human brain [6]. In addition to mechanically depolarized or structurally impaired neurons, nutritional and oxygen deprivation also contributes to this dramatic increase in glutamate concentration, leading to an inversion of astrocyte glutamate transporter [7] and impaired glutamate uptake by presynaptic neurons [8].
Subsequently, glutamate causes Ca2+-dependent neurotoxicity due to prolonged activation of NMDA-type ionotropic glutamate receptors, which have a high permeability to extracellular Ca2+ and Na+. A sharp increase in intracellular free Ca2+ concentration ([Ca2+]i) activates various mechanisms of cell death, leading to additional loss of neurons long after the primary damage. Therefore, a decrease in [Ca2+]i after mechanical damage to neurons in vitro correlates with a decrease in neuronal damage and an increase in their viability. A reproduction of such results in general indicates the neuroprotective activity of the studied preparations [9].
The increase in intracellular Na concentration near the NMDA channel also plays a substantial role in the pathophysiology of TBI. This leads to a massive membrane depolarization and increase in H2O retention, direct water movement into the intracellular compartment, and, consequently, cytotoxic edema. At the same time, Na+-dependent depolarization causes Cl− influx, which only aggravates neuronal swelling and, consequently, cell death [10]. Another factor increasing intracellular Na+ and Cl− is NKCC1, which has been shown [11] to be upregulated after TBI. The main consequence of cytotoxic edema, cell death, was previously attributed to its positive effect on intracellular Ca2+ accumulation [12]. However, another study [10] suggested that cell death in neuronal edema occurs even in the absence of dramatic [Ca2+] changes, and can be prevented by blocking depolarization-dependent Cl- influx.
There are various in vitro models of mechanical neurotrauma. Models of acute penetrating impact allow the reproduction of the process of neuronal damage using a flexible plastic pipette tip [13], a circular slicer, or a razor [14][15]. These and other models [16][17] suggest an acute disruption of cell integrity, exposing neighboring neurons to excitotoxic effects and causing glial cells activation. A limitation of all these models is the lack of standardized parameters for the dynamics of mechanical injury and assessment of its severity, primarily based on the number of damaged cells, which introduces great uncertainty in the interpretation of results.
2. The Pathogenesis of TBI
The pathological processes underlying the pathogenesis of TBI are schematically shown in Figure 1. A progressive increase in pathological phenomena after TBI is observed in 50% of the victims. The consequences of TBI include a number of neurological and psychiatric abnormalities that can be detected 2 to 10 years after injury in 90% of patients. These disorders usually determine the social disadaptation of patients, often leading to temporary disability, often turning into permanent disability [18]. In craniocerebral trauma, there are disorders of cerebral circulation, cerebrospinal fluid circulation, and permeability of the blood–brain barrier. Of course, the severity of pathological changes in the brain varies with different degrees of TBI severity [19]. Due to excessive water influx into brain cells and intercellular space, edema of the brain develops, which causes an increase in intracranial pressure. The processes of brain displacement and compression develop. This, in turn, causes further deterioration of blood circulation, metabolism, and functional activity of the brain. An adverse secondary factor of brain damage is its hypoxia due to respiratory or circulatory disturbances [18][20][21]. Thus, post-traumatic syndrome is a symptom complex that includes dizziness, headache, difficulties with concentration, nervousness, insomnia, and personality disorders.
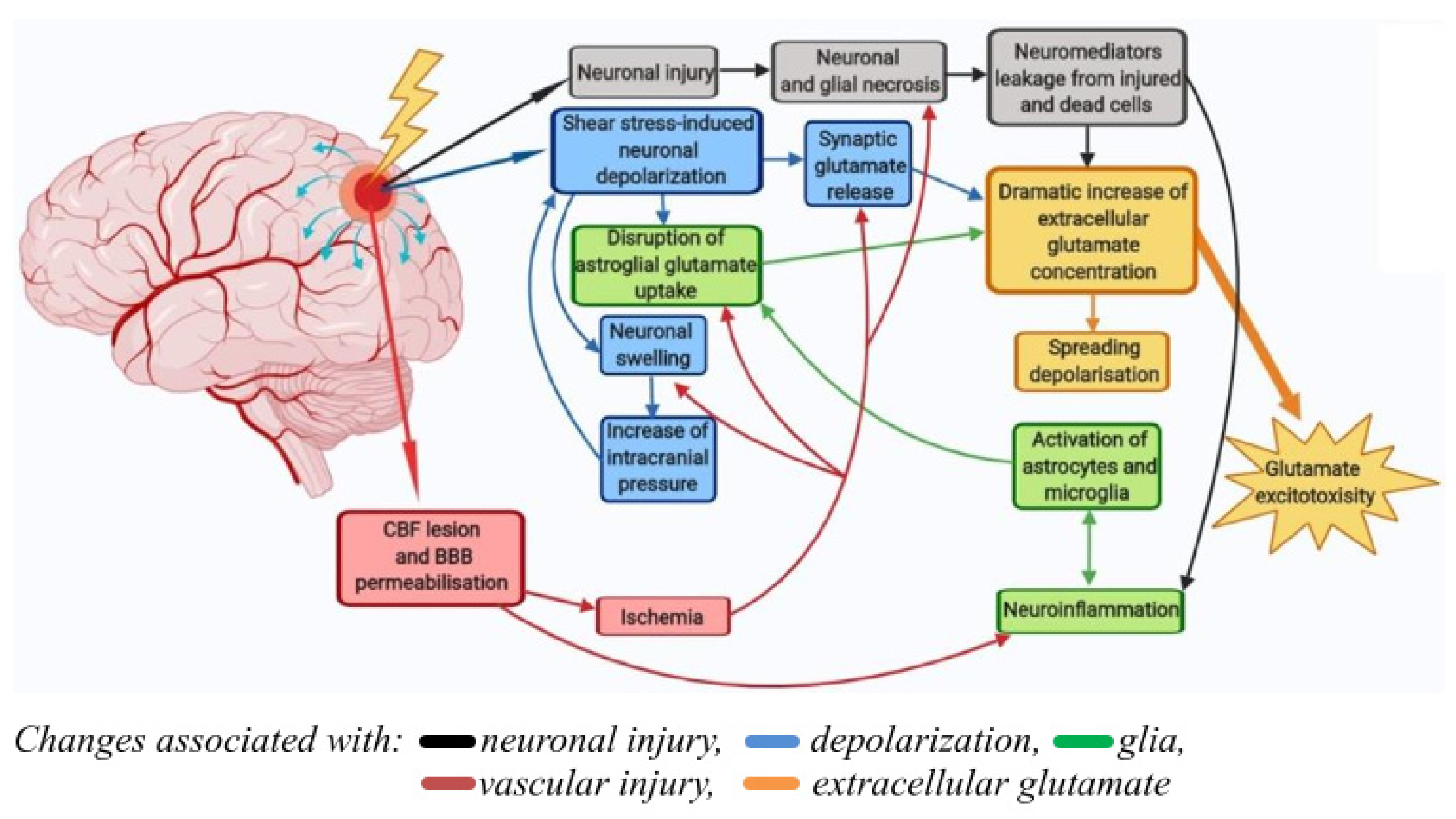
Figure 1. Pathological processes underlying the pathogenesis of traumatic brain injury. Primary damage at TBI is due to the mechanical impact itself (force, degree of penetration, and area of damage) and develops in the first few seconds after the trauma (black arrows). Secondary damage is longer-term (hours-days) and is mediated by primary damage mechanisms. They include pathological depolarization due to shear stress (blue arrows), changes in CBF dynamics, as well as neuro-inflammatory processes caused by glial and immune cells coming from the bloodstream during BBB permeabilization (green and red arrows). The delayed neuronal death caused by TBI is due to the excitotoxicity of glutamate (yellow arrow).
There is a limited number of models that allow the assessment of the effect of mechanical trauma on Ca2+ homeostasis and the viability of primary cortical neurons [22]. Moreover, there are still no studies that have examined the acute (first seconds, minutes) and delayed (several days after mechanical injury) effects of mechanical trauma on cellular viability and the dynamic of changes in [Ca2+]i and mitochondrial potential (ΔΨm) of neurons. The closest study is the one in which a stretch injury was applied to neurons and then their excitability was assessed using a patch clamp 30–60 min after the injury [23]. Another study also used a similar model of stretch injury, but in it, the viability and metabolic functions of neurons were recorded 24 h after the impact [24].
3. Current Insights
Several parameters influence the magnitude of the integral fluorescence response during glutamate administration. The most important of them is the rate of the DCD onset, i.e., the multistep increase in [Ca2+]i, which is mirrored by the rapid increase in the Fura-FF fluorescent signal. The maximum fluorescence level in this phase of the experiment also depends on the efficiency of extra mitochondrial systems responsible for Ca2+ removal from the cytoplasm, such as membrane transporters PMCA, plasma NCX. The magnitude of the integral fluorescent response and the degree of [Ca2+]i recovery during EGTA solution administration are additional parameters to assess the degree of Ca2+-homeostasis stability. Replacing the glutamate solution with EGTA solution not only reduces the deleterious activation of NMDA receptors activation, but also changes the Ca2+ ion concentration gradient between the cytoplasm and the external environment in the opposite direction, which allows cells that have not completely lost their homeostatic potential to remove Ca2+ from the cytoplasm. In Ca2+-free buffer, neurons from mechanically injured cultures poorly recovered the initial [Ca2+]i compared to controls.
It is known that DCD is associated with permeabilization of the inner mitochondrial membrane. This phenomenon is accompanied by mitochondrial depolarization, which coincides in time with the increase in cytoplasmic Ca2+ concentration [25]. All differences between the groups were observed at the moment of repolarization of the inner mitochondrial membrane during the post-glutamate period. This may indicate that pathological processes triggered by mechanical injury affect the dynamics of the transition between the open and closed state of the mitochondrial permeability transition pore, as well as the functionality of the electron transport chain. It is known that [Ca2+]i recovery can be promoted by the closure of mitochondrial permeability transition pores [26], which leads to the repolarization of the inner mitochondrial membrane and the recovery of the MCU providing the calcium return to the mitochondria.
It has been shown that the significant [Ca2+]i spike and synchronous depolarization of mitochondria could be caused by NMDA channel activation, as well as by other, yet unidentified mechanisms. This conclusion is based on the fact that MK-801 blocked the high spike in [Ca2+]i and the sharp drop in ΔΨm only in a part of the neurons, whereas removing Ca2+ from the buffer abolished large changes in [Ca2+]i and ΔΨm in all cells. In a mature neuroglial culture, the length of axons can reach hundreds of micrometers [27], while the length of astrocyte dendrites does not exceed 20–30 microns [28].
Thus, mechanical injury causes strong and abrupt changes in [Ca2+]i and ΔΨm in cells close to the damage zone. These changes are comparable with those caused by neurotoxic doses of glutamate. In some neurons, these changes are indeed caused by stimulation of Glu-directed NMDA receptors, whereas in other cells, ATP serves as the signal molecule. On the other hand, the maximal level of [Ca2+]i in the glutamate washout phase depends on the efficiency of extra mitochondrial systems for Ca2+ removal from the cytoplasm, such as PMCA, and plasma NCX. Their efficiency is dependent directly or indirectly on the ATP concentration in the cytosol, which is determined by the intensity of cellular respiration. An increase in the [Ca2+]i during exposure to glutamate in neurons from injured cultures compared to control neurons may indicate impaired ATP synthesis. Thus, mechanical trauma disrupts Ca2+ homeostasis systems in adjacent neurons. Impairment of calcium homeostasis in surviving neurons after TBI is not immediately apparent and may be the cause of post-traumatic syndrome. In this case, a protective therapy against [Ca2+]i increase or ΔΨm loss under glutamate excitotoxicity may be promising.
4. Conclusions
A model was developed of mechanical neurotrauma that allows us to measure the dynamics of [Ca2+]i and ΔΨm both in the first seconds, minutes, and several days after injury [29]. NMDA receptor ion channels serve as the main pathway for Ca2+ entry into neurons in the event of mechanical damage to a neuronal culture. Ca2+ overload of mitochondria and a prolonged decrease in ΔΨm can cause delayed neuronal death and thus play a key role in the development of the post-traumatic syndrome. The process of the neuroglial culture repair were also able to be visualized, where neurons were the first in the damaged area and were followed by astrocytes. Nevertheless, the viability of neurons inevitably decreased two weeks after injury. This effect may be attributed to the long-term metabolic dysregulation of neurons caused by trauma. Therefore, the use of neuroprotective compounds would allow us to test this hypothesis.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23073858
References
- Hiebert, J.B.; Shen, Q.; Thimmesch, A.R.; Pierce, J.D. Traumatic Brain Injury and Mitochondrial Dysfunction. Am. J. Med. Sci. 2015, 350, 132–138.
- Werner, C.; Engelhard, K. Pathophysiology of Traumatic Brain Injury. BJA Br. J. Anaesth. 2007, 99, 4–9.
- Fujikawa, D.G. The Role of Excitotoxic Programmed Necrosis in Acute Brain Injury. Comput. Struct. Biotechnol. J. 2015, 13, 212–221.
- Andriessen, T.M.J.C.; Jacobs, B.; Vos, P.E. Clinical Characteristics and Pathophysiological Mechanisms of Focal and Diffuse Traumatic Brain Injury. J. Cell. Mol. Med. 2010, 14, 2381–2392.
- Arundine, M.; Tymianski, M. Molecular Mechanisms of Glutamate-Dependent Neurodegeneration in Ischemia and Traumatic Brain Injury. Cell. Mol. Life Sci. CMLS 2004, 61, 657–668.
- Bullock, R.; Zauner, A.; Woodward, J.J.; Myseros, J.; Choi, S.C.; Ward, J.D.; Marmarou, A.; Young, H.F. Factors Affecting Excitatory Amino Acid Release Following Severe Human Head Injury. J. Neurosurg. 1998, 89, 507–518.
- Yi, J.-H.; Hazell, A.S. Excitotoxic Mechanisms and the Role of Astrocytic Glutamate Transporters in Traumatic Brain Injury. Neurochem. Int. 2006, 48, 394–403.
- Dorsett, C.R.; McGuire, J.L.; Niedzielko, T.L.; DePasquale, E.A.K.; Meller, J.; Floyd, C.L.; McCullumsmith, R.E. Traumatic Brain Injury Induces Alterations in Cortical Glutamate Uptake without a Reduction in Glutamate Transporter-1 Protein Expression. J. Neurotrauma 2017, 34, 220–234.
- Lapanantasin, S.; Chongthammakun, S.; Floyd, C.L.; Berman, R.F. Effects of 17β-estradiol on Intracellular Calcium Changes and Neuronal Survival after Mechanical Strain Injury in Neuronal–Glial Cultures. Synapse 2006, 60, 406–410.
- Rungta, R.L.; Choi, H.B.; Tyson, J.R.; Malik, A.; Dissing-Olesen, L.; Lin, P.J.C.; Cain, S.M.; Cullis, P.R.; Snutch, T.P.; Macvicar, B.A. The Cellular Mechanisms of Neuronal Swelling Underlying Cytotoxic Edema. Cell 2015, 161, 610–621.
- Lu, K.T.; Cheng, N.C.; Wu, C.Y.; Yang, Y.L. NKCC1-Mediated Traumatic Brain Injury-Induced Brain Edema and Neuron Death via Raf/MEK/MAPK Cascade. Crit. Care Med. 2008, 36, 917–922.
- Kimelberg, H.K. Water Homeostasis in the Brain: Basic Concepts. Neuroscience 2004, 129, 851–860.
- Tecoma, E.S.; Monyer, H.; Goldberg, M.P.; Choi, D.W. Traumatic Neuronal Injury in Vitro Is Attenuated by NMDA Antagonists. Neuron 1989, 2, 1541–1545.
- Mukhin, A.G.; Ivanova, S.A.; Knoblach, S.M.; Faden, A.I. New in Vitro Model of Traumatic Neuronal Injury: Evaluation of Secondary Injury and Glutamate Receptor-Mediated Neurotoxicity. J. Neurotrauma 1997, 14, 651–663.
- Mukhin, A.G.; Ivanova, S.A.; Allen, J.W.; Faden, A.I. Mechanical Injury to Neuronal/Glial Cultures in Microplates: Role of NMDA Receptors and PH in Secondary Neuronal Cell Death. J. Neurosci. Res. 1998, 51, 748–758.
- Laskowski, A.; Schmidt, W.; Dinkel, K.; Martínez-Sánchez, M.; Reymann, K.G. BFGF and EGF Modulate Trauma-Induced Proliferation and Neurogenesis in Juvenile Organotypic Hippocampal Slice Cultures. Brain Res. 2005, 1037, 78–89.
- Allen, J.W.; Knoblach, S.M.; Faden, A.I. Combined Mechanical Trauma and Metabolic Impairment in Vitro Induces NMDA Receptor-dependent Neuronal Cell Death and Caspase-3-dependent Apoptosis. FASEB J. 1999, 13, 1875–1882.
- Hawley, C. Management of Minor Head Injury in Adults. Emerg. Nurse 2010, 18, 20–24.
- Dikmen, S.; Machamer, J.; Temkin, N. Mild Traumatic Brain Injury: Longitudinal Study of Cognition, Functional Status, and Post-Traumatic Symptoms. J. Neurotrauma 2017, 34, 1524–1530.
- Theeler, B.; Lucas, S.; Riechers, R.G.; Ruff, R.L. Post-traumatic Headaches in Civilians and Military Personnel: A Comparative, Clinical Review. Headache J. Head Face Pain 2013, 53, 881–900.
- Marshman, L.A.G.; Jakabek, D.; Hennessy, M.; Quirk, F.; Guazzo, E.P. Post-Traumatic Amnesia. J. Clin. Neurosci. 2013, 20, 1475–1481.
- Li, Y.; Li, C.; Gan, C.; Zhao, K.; Chen, J.; Song, J.; Lei, T. A Precise, Controllable in Vitro Model for Diffuse Axonal Injury through Uniaxial Stretch Injury. Front. Neurosci. 2019, 13, 1063.
- Magou, G.C.; Pfister, B.J.; Berlin, J.R. Effect of Acute Stretch Injury on Action Potential and Network Activity of Rat Neocortical Neurons in Culture. Brain Res. 2015, 1624, 525–535.
- Rosas-Hernandez, H.; Burks, S.M.; Cuevas, E.; Ali, S.F. Stretch-Induced Deformation as a Model to Study Dopaminergic Dysfunction in Traumatic Brain Injury. Neurochem. Res. 2019, 44, 2546–2555.
- Khodorov, B.I.; Storozhevykh, T.P.; Surin, A.M.; Yuryavichyus, A.I.; Sorokina, E.G.; Borodin, A.V.; Vinskaya, N.P.; Khaspekov, L.G.; Pinelis, V.G. The Leading Role of Mitochondrial Depolarization in the Mechanism of Glutamate-Induced Disruptions in Ca2+ Homeostasis. Neurosci. Behav. Physiol. 2002, 32, 541–547.
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155.
- Waseem, T.; Mukhtarov, M.; Buldakova, S.; Medina, I.; Bregestovski, P. Genetically Encoded Cl-Sensor as a Tool for Monitoring of Cl-Dependent Processes in Small Neuronal Compartments. J. Neurosci. Methods 2010, 193, 14–23.
- Verkhratsky, A.; Olabarria, M.; Noristani, H.N.; Yeh, C.-Y.; Rodriguez, J.J. Astrocytes in Alzheimer’s Disease. Neurotherapeutics 2010, 7, 399–412.
- Zanda Bakaeva; Mikhail Goncharov; Irina Krasilnikova; Arina Zgodova; Daniil Frolov; Ekaterina Grebenik; Peter Timashev; Vsevolod Pinelis; Alexander Surin; Acute and Delayed Effects of Mechanical Injury on Calcium Homeostasis and Mitochondrial Potential of Primary Neuroglial Cell Culture: Potential Causal Contributions to Post-Traumatic Syndrome. International Journal of Molecular Sciences 2022, 23, 3858, 10.3390/ijms23073858.
This entry is offline, you can click here to edit this entry!