Aging is a broad process that occurs as a time-dependent functional decline and tissue degeneration in living organisms. On a smaller scale, aging also exists within organs, tissues, and cells. As the smallest functional unit in living organisms, cells “age” by reaching senescence where proliferation stops. Such cellular senescence is achieved through replicative stress, telomere erosion and stem cell exhaustion. It has been shown that cellular senescence is key to tissue degradation and cell death in aging-related diseases (ARD). However, senescent cells constitute only a small percentage of total cells in the body, and they are resistant to death during aging. This suggests that ARD may involve interaction of senescent cells with non-senescent cells, resulting in senescence-triggered death of non-senescent somatic cells and tissue degeneration in aging organs.
1. Introduction
Aging is often defined as progressive physiological degeneration, leading to impaired function and increased vulnerability to death. It has become an increasingly burdensome challenge due to the rapid growth of the aging human population. A 2015 report from the UN estimates that by 2050, there will be more than 2.1 billion people over the age of 60 due to an increased birth rate, improved healthcare, and better living conditions
[1]. Several diseases are strongly correlated to advanced age, including Alzheimer’s disease, cardiovascular disease, osteoarthritis, diabetes and cancer; all roughly double in incidence every five years after age 60
[2][3]. So far, many ARDs lack effective cures
[4].
The decline in organ and tissue function of ARD reflects the underlying molecular aberrations driving the cellular aging process
[2]. Nine causative mechanisms of aging have been proposed with the criteria that each occurs during normal aging, accelerates aging when experimentally aggravated, and decelerates aging when experimentally ameliorated. These hallmarks include cellular senescence, stem cell exhaustion, altered intercellular communication, genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, and mitochondrial dysfunction
[4]. Cellular senescence is not only a leading cause of aging, but also the cellular basis of the other causative mechanisms of aging. Although remarkable progress has been made in understanding cell senescence since it was proposed by Hayflick sixty years ago
[5], it is still a work in progress and a hot area of research currently. Still unclear is how cell senescence contributes to the major phenotypes of ARDs including fibrosis, soft tissue ossification (calcification), inflammaging (chronic low-grade inflammation), cell death and tissue degeneration. Uncovering the molecular and cellular mechanisms of cell senescence will have a profound impact on the understanding of aging and developing interventions of ARDs.
Aged tissues and organs contain a complex mixture of quiescent, proliferative, and senescent cell populations
[6]. The cell numbers, types and functions change dynamically during aging, which are often thought to contribute to disease progression. Cellular senescence is closely associated with such processes.
2. Cell Senescence Is Cell-Autonomous: Telomere Shortening, Replicative Stress and SASP Manifestation
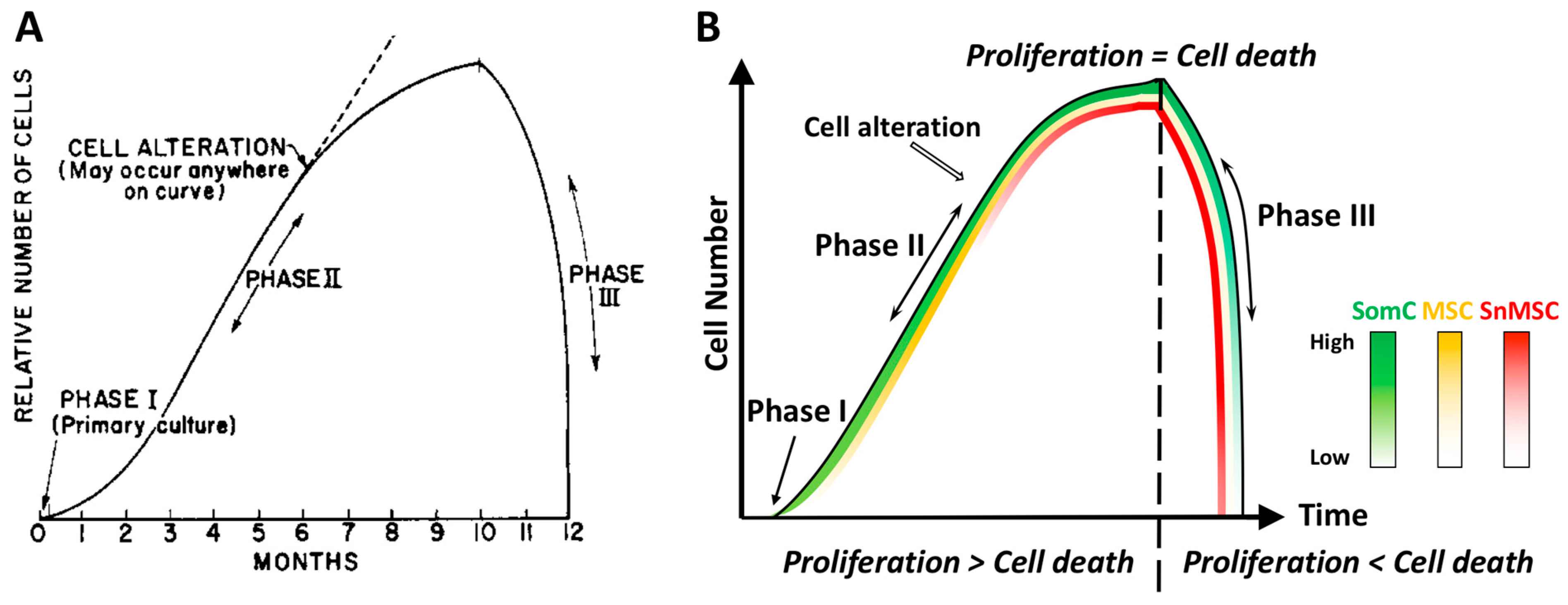
Almost 60 years ago, Leonard Hayflick and Paul Moorhead first reported that primary human cells derived from embryonic tissues exhibited limitations while dividing
[5]. These fibroblasts could only divide for 40–60 times in cell culture before entering senescence. Hayflick defined the different stages of cell culture into three phases (
Figure 1). Phase I is the original primary culture, where cells divide to cover the surface at a relatively slow rate. Phase II represents the period when robust cell division occurs. Continuous subcultures of the same batch of cells leads to Phase III, when cells not only stopped growth but also died in a short period of time. Hayflick postulated that cell alterations must have occurred during cell growth in Phase II, which led to cell growth arrest and death in Phase III. He called this phenomenon cell senescence and further postulated that it could be a mechanism to counter unlimited cell proliferation in cancer. This phenomenon of cell growth arrest is later defined as Hayflick Limit due to replicative senescence (
Figure 1A). In a discovery that led to a Nobel prize on telomerase, it was found that the primary cause for Hayflick Limit, or replicative senescence, is telomere shortening
[7][8].
Telomeres are non-coding regions at the end of chromosomes, which consist of thousands of the same sequence repeats
[7][8]. Due to the nature of lagging-strand synthesis, DNA polymerase cannot replicate the full 3′ end of duplex DNA
[9]. This leads to the loss of small segments of DNA within telomeres. After a certain number of divisions, the telomeres reach a critical length where cells become senescent. Thus, telomere shortening drives the activation of cell senescence mechanisms. However, cell growth arrest by itself may not be harmful to our body, since most somatic cells in a mature adult organ are post-mitotic or divide very slowly. The detrimental effects of cell senescence were not clear until the discovery of the senescence-associated secretory phenotype (SASP)
[10].
The discovery of SASP demonstrated that, even though they no longer proliferate, senescent cells remain metabolically active and secrete a broad spectrum of factors, including cytokines, chemokines, proteases, growth factors, receptors and ligands, and extracellular matrix components
[10]. These SASP factors can cause autocrine and paracrine inflammatory responses. Some of the SASP factors, such as interleukin-1 (IL-1), IL-6, IL-8, TNF-α, and matrix metalloproteinase1 (MMP1)
[11], contribute to persistent chronic inflammation in the microenvironment, which is often associated with multiple aging-related phenotypes
[12][13]. In addition to SASP, senescent cells also influence the microenvironment through cell the secretome including microvesicles (MVs). Typically, MVs function as a key component of the cell secretome in immunomodulatory regulation and tumor growth inhibition
[14][15]. However, senescent cells change the composition of MVs to negatively impact the microenvironment. Exosomes, an important type of MVs, contain biological active molecules including cytokines, growth factors, and regulatory miRNA
[16]. Senescence greatly alters the composition of their exosomes, especially the miRNA content. RNAseq analysis of senescent mesenchymal stromal cell (MSC) MVs identified many highly expressed genes associated with ARDs
[17].
3. Cell Senescence-Associated Apoptosis and Degeneration Is Senescent Cell Non-Autonomous: The SACTAI Mechanism for Aging
The Hayflick phenomenon included Phase III when cell number rapidly declines after reaching the Hayflick Limit (cell senescence) (
Figure 1A). While the mechanism for the increased cell number to result in cell senescence in Phase I and II has been elucidated, the mechanism for senescence-associated cell death in Phase III was not addressed or even ignored. Cell death (apoptosis) and degeneration are also hallmarks of ARDs in vivo. However, senescent cells are resistant to apoptosis, which is a characteristic of cell senescence
[18][19]. The significant decrease in cell number in aging tissues in vivo and in primary cell cultures in vitro is likely not due to apoptosis of senescent cells, but other non-senescent cells. Thus, while cell senescence is a cell autonomous process, cell senescence-associated apoptosis and degeneration may not be.
Based on RNAseq of human aging cartilage tissues and primary chondrocyte culture passage experiments, a mechanism of Senescence-Associated Cell Transition and Interaction (SACTAI) was proposed
[20]. SACTAI is induced by repeated activation of somatic cell (SomC) replication in response to stress signals during aging or after injury. This causes cell type transitions from differentiated SomCs to MSCs by de-differentiation, and further into senescent MSC (snMSC) due to replicative stress (
Figure 1B).
Although it was thought for a long time that chondrocytes were the only cell type in cartilage tissue, evidence for the existence of different cell types has been shown in aging human articular cartilage in recent years
[21][22][23][24][25]. Further, the percentage of progenitor/MSC was increased in osteoarthritis (OA) cartilage, suggesting aging-induced cell heterogeneity
[22][26].
4. Two-Way Communications between Senescent and Non-Senescent Cells: Altered Intercellular Signaling Mechanisms during Aging
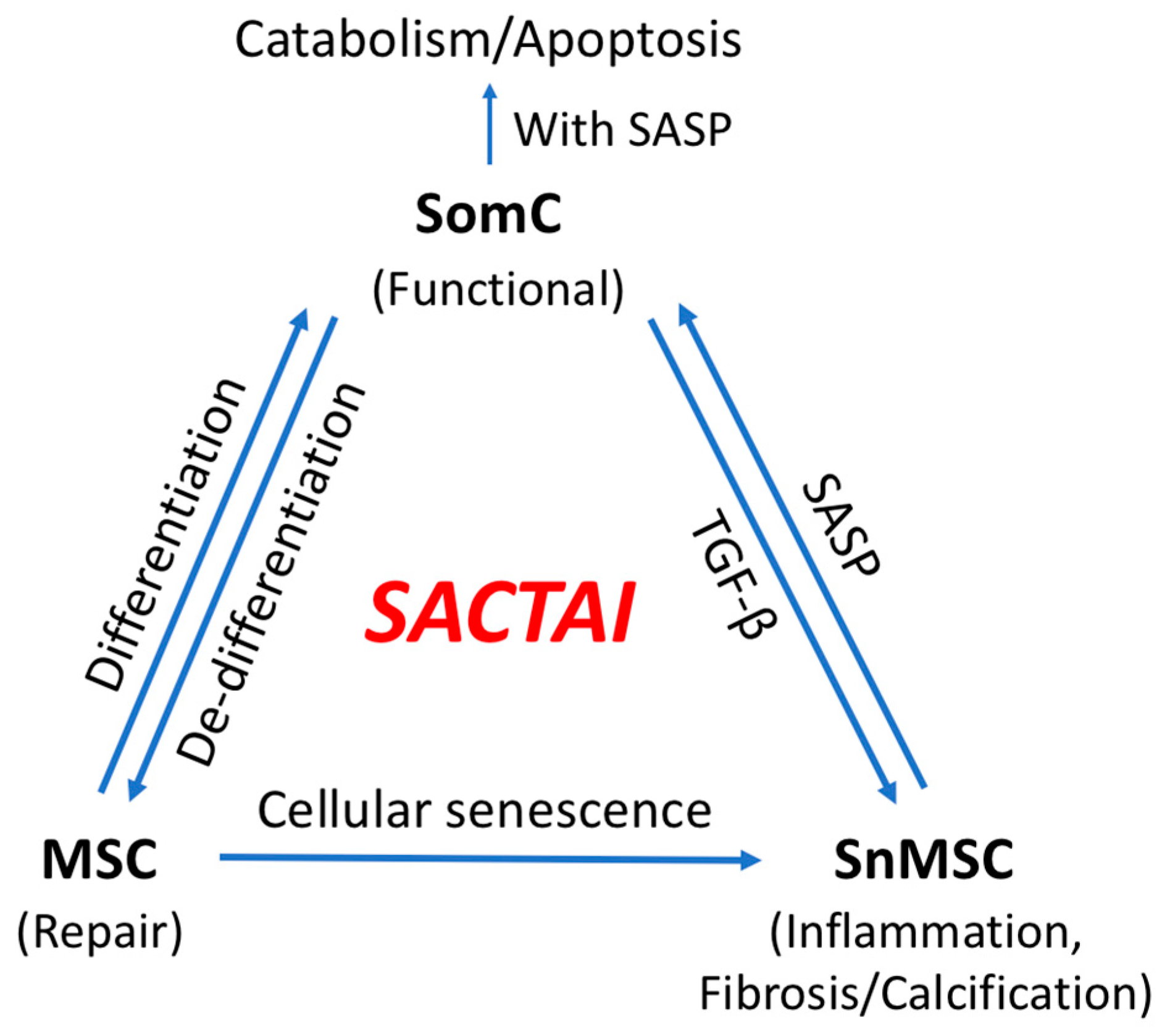
An important consequence of senescence-associated cell transition is enabling heterotypic cell interaction between SnMSCs and non-senescent SomC. SASPs appear to be major signaling molecules mediating the SACTAI, as shown by recent publications
[27][28]. The interaction between senescent and non-senescent cells is a two-way communication. The first is signaling from senescent cells to non-senescent cells (
Figure 2). snMSC acquire a pro-inflammatory phenotype and secrete cytokine IL-1β due to cell transition and senescence. On the other hand, chondrocytes are the main recipient cells of inflammatory signaling by expressing IL-1R
[20]. In addition to cytokine signaling, a recent study also identified Sonic Hedgehog (SHH) to mediate senescence signaling from snMSC to SomC
[27]. SHH is known to play an important role during skeletal development and homeostasis
[29][30]. SHH is expressed by NCSCs and increased in OA-MSCs during aging
[27].
5. SACTAI in Adult Tissues
In a typical tissue, the cells may be of the same type or of multiple types. The majority of cells are very distinctive and perform specific functions. These SomCs are mature and terminally differentiated cells, which usually carry out specialized tasks in the body. MSCs can divide and differentiate, are responsible for tissue morphogenesis during development and growth and repair during the adult stage. The MSCs in the adult stage are the focus here. Together SomCs, MSCs, and SnMSCs ensure the proper functions of an organ and maintain tissue homeostasis.
5.1. Dedifferentiation: SomC to MSC Transition
In adult tissue, the most common cellular state is that of terminally differentiated SomCs. A plethora of different terminally differentiated SomCs comprises each adult tissue, with each cell type taking on a unique function within the tissue. These SomCs are often thought to have permanently exited the cell cycle and are incapable of replicating or replacing themselves
[31]. Maintaining a stable cellular identity is crucial for normal tissue function. Such stability is achieved through epigenetic regulation
[32]. However, cell identity can be altered under experimental conditions. For example, John Gurdon performed nuclear transplantation on mature adult cells and converted them into cells with properties of a fertilized egg
[33].
5.2. MSC Senescence
MSCs from development are a small population of cells that reside in the tissue throughout most postnatal life
[34]. They are undifferentiated cells and give rise to a limited number of mature cell types that play roles in the functions of a tissue
[32][35]. These adult stem cells are self-renewing, clonogenic, and multipotent in nature. Thus, they are critical to maintaining the tissue homeostasis. Adult stem cells are generally in a predominantly quiescent state for long-term maintenance of tissue functions
[36][37]. Upon the loss of functional cells either through aging or injury to the tissue, they are activated to proliferate and differentiate into the required cell types. Therefore, adult stem cells play a key role in tissue repair. These MSCs are found in a variety of tissues, including bone marrow, adipose tissue, cartilage, synovial fluid, brain, skin, lung, dental pulp, umbilical cord blood, and placenta
[38][39][40][41][42]. They can differentiate into multiple cell lineages, such as bone, cartilage, adipose, and neuronal cells
[38]. Thus, MSCs have emerged as powerful tools for therapeutic applications in tissue engineering and regenerative medicine
[43]. Applications of MSC-based therapy have been tested in different diseases, such as graft-vs.-host disease (GVHD), diabetes mellitus (DM), Crohn’s disease (CD), multiple sclerosis (MS), myocardial infarction (MI), etc.
[43][44].
5.3. BM-MSC Senescence
Bone marrow-derived MSCs (BM-MSCs) are the most investigated MSCs. They play a role in the regulation of the local microvascular network, differentiation to osteoblasts, and maintain the hematopoietic microenvironment
[45]. Multiple studies have shown cell type transition in BM-MSCs. Maderia et al. performed ex-vivo cultivation of primary human BM-MSCs from healthy donors
[46].
6. SACTAI: A Mechanism of Balancing Tissue Repair and Degeneration
In healthy tissues/organs, cellular senescence is not only a well-known hallmark of aging, but also an effective mechanism to prevent proliferation of damaged cells. Stem cell senescence is a cellular response to endogenous or exogenous stresses, including accumulated stresses through aging. During the early stages of life, it is a protective mechanism to limit proliferation and function of aberrant cells
[47]. However, cell senescence is not always good, especially in aged tissues. The accumulation of senescent cells during aging contributes to disease pathogenesis and exhibits negative effects on non-senescent cells
[47]. Such changes from beneficial to detrimental effects can be explained by SACTAI. In a young adult, the tissue environment contains mostly differentiated SomCs with specific roles to ensure proper function of the organ. However, a small pool of quiescent MSCs is left in the tissue as a backup for repair. When a young individual encounters a stress and requires damage repair, MSCs exit the quiescent state and resume their abilities for self-renewal and differentiation. Such robust cell division correlates with Hayflick’s Phase II before the SnMSC is induced (
Figure 1). Thus, SomCs and MSCs maintain a balance to carry out tissue regeneration without reaching their replication capacity. In a young adult tissue, tissue repair is balanced with tissue degeneration.
Senescence-associated cell transition is similar to a well-studied cell transition pathway, epithelial to mesenchymal transition (EMT). EMT is also reversible, known as a mesenchymal-epithelial transition (MET)
[48]. During embryonic development, the roles of EMT/MET are indispensable as they contribute to the formation and function of developing organs
[49]. However, EMT/MET transitions in adult life often play roles in ARDs including organ fibrosis and cancer
[50][51].
7. SACTAI in Age-Related Diseases
ARDs are triggered by the stress signals that result in cell senescence including oxidative, irradiation, chemical, mechanical, inflammatory, and replicative stress
[47]. The concept of SACTAI can be applied to cells in different tissues during aging. Both in vivo and in vitro data have indicated the cell type transitions and interactions in different tissues, and how they contribute to ARDs (
Figure 2). It provides a general mechanism for aging and aging-associated tissue repair and degeneration.
7.1. Physical Dysfunction
Physical function declines with aging, causing disability, increased health expenditure and mortality
[52][53]. Obesity and physical inactivity are often associated with physical dysfunction
[54][55]. The cellular pathogenesis of age-related physical dysfunction is not fully understood. There are currently no mechanism-based interventions for improving physical function in the elderly. Adipose tissue is the major source of obesity and inactivity-related inflammation
[54]. It contains a population of MSCs with multilineage differentiation along adipogenic, osteogenic, chondrogenic, and neurogenic lineages.
7.2. Cardiovascular Disease
Cardiovascular disease is the most common cause of death in elderly people. It includes chronic ischemic heart disease, congestive heart failure and arrhythmia
[3]. Adult human heart is known to harbor a small population of resident stem cells, also known as cardiac stem and progenitor cells (CPCs)
[56][57][58]. These CPCs enable the adult heart to self-renew over the human lifespan
[59]. The function of CPCs decreases with aging, leading to cardiovascular disease
[60][61].
7.3. Neurodegenerative Disorder
Neurodegenerative disorders are characterized by progressive neuronal death and loss of specific neuronal cell populations. Axonal degeneration takes place as a consequence of normal aging. Thus, age constitutes the main risk factor for its pathogenesis. Neurodegenerative disorders include Alzheimer’s disease, Parkinson’s disease, multiple sclerosis (MS), Huntington’s disease, and amyotrophic lateral sclerosis
[62][63]. It has been shown that neural progenitor cells (NPCs) play important roles in neurodegenerative disorders. NPCs have the capacity to differentiate into neurons, astrocytes and oligodendrocytes to replace the damaged cells in adult brain
[63].
7.4. Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is a chronic interstitial lung disease defined by a progressive and irreversible loss of lung function through scar tissue accumulation
[64][65][66]. There is an increase in cell senescence markers in lung fibroblasts from IPF patients
[66][67][68][69].
7.5. Bone and Joint Degeneration
OA is a common age-associated disease in the knee joint. It is a consequence of progressive age-induced senescence in the cartilage, leading to changes such as articular cartilage degradation, osteophyte formation, and subchondral bone sclerosis
[70][71][72]. Repair of the joint cartilage relies on chondrocytes and resident MSCs. These MSCs are often found in articular cartilage, sub-chondral bone marrow and synovial tissue
[73][74][75][76].
8. Conclusions
8.1. Recent Finding
Recent genomic analysis reveals a remarkable heterogeneity of cell types during aging. Such cell heterogeneity gives rise to not only senescent cells but also other types of cells including progenitor and stromal cells. Adult MSCs constitute a small percentage of cells responsible for repair upon tissue damage. The increase in senescent cells is tightly associated with repeated activation of adult MSCs, where they reach replication capacity and become senescent. In response to stress signals, differentiated SomCs may lose their identity and de-differentiate into MSC-like cells for repair. Such epigenetically re-programmed MSCs are subject to cell senescence triggered by replicative, mechanical, and inflammatory stress signals. Although in small numbers, senescent MSCs manifest SASP, spread inflammation, and signal surrounding somatic cells in the tissue microenvironment. Thus, senescent MSCs may accumulate during aging by cell proliferation, transition, and senescence, and accelerate catabolism and death of somatic cells through cell interactions. Important signaling molecules mediating the SACTAI process include pro-inflammatory cytokine IL-1β, IL-6, IL-8, growth factor TGF-β, and morphogen Sonic Hedgehog, which at least partially overlap with SASPs.
8.2. Summary
SACTAI is a proposed two-step mechanism for aging-associated tissue degeneration and somatic cell death. In the first step, a few adult SomCs, in response to mechanical, inflammatory, or replicative stress signals, undergo proliferation, MSC transition and senescence, resulting in senescent MSCs (snMSCs). This cell transition and senescence process results in a heterogenous cell population, which enables heterotypic cell interactions with each other. During the second step, snMSCs interacts with SomCs via SASPs. Such cell senescence-associated signaling contributes to cell death and tissue degeneration in ARD. The newly discovered SomC transition to snMSC during aging may explain the fibrosis, abnormal ossification (calcification), and inflammaging phenotypes often associated with aging tissues. The identification of the multi-step mechanism of SACTAI provides an opportunity to develop potential drugs to intervene during different stages of ARD pathogenesis.
8.3. Implication
SACTAI provides a general mechanism for aging in different types of tissues. With increasing adult stromal cells during aging-associated cell heterogeneity, cell type transition is crucial to understanding the cellular processes leading to cell senescence, fibrosis, inflammation, and degradation. It is a dynamic and gradual process based on the balance between regeneration and degeneration. As different cell types (SomCs, MSCs, and SnMSCs) co-exist in the same microenvironment, cell transition and interaction occur both spatially and temporally. The mesenchymal cell transition and senescence may not only account for fibrosis, abnormal ossification, and inflammaging phenotypes of ARDs, but also provide key intervention points for treatment. Future studies on the mechanism of SACTAI in different tissues will shed light on new interventions and treatments of ARDs.
This entry is adapted from the peer-reviewed paper 10.3390/cells11071089