There are a lot of lignocellulolytic bacteria in rumen. These bacteria secrete carbohydrate-active enzymes (CAZymes) to help digest complex and recalcitrant lignocellulose material of roughage in the host. In comparison with bacteria, anaerobic fungi not only possess physical penetration effect on plant tissue, but also secret a group of highly specialized CAZymes, including GH10, GH11, GH6, GH45, GH5, GH43, CE1, CE4, and and so on with higher abundance than bacteria during the enzymatic process
[35].
2.1. Physical Degradation with Fungal Rhizoids
Anaerobic fungi use flagella as their locomotor organ, and the chemotaxis of moving fungal zoospores to soluble sugars enables them to move quickly ahead of other rumen microorganisms and colonize on the ingested plant tissue
[36]. It may provide a unique advantage for fewer anaerobic fungi to compete for nutrients in the complex rumen environment
[37]. When they touch the surface of the plant, the zoospores spread out their flagella to form cysts. During the formation and growth of cysts, the rhizoids originate from the side of the cell opposite the insertion of the flagella, and is polar or lateral in position
[2], and these develop into a highly branched rhizoid system. The rhizoid system of the anaerobic fungi then penetrates the plant tissue and destroys its structure. This first step is physical degradation, which exposes a larger area for interaction between digestive enzymes and fibrous tissue, and releases the plant cell contents for utilization by itself (fungi) and other rumen microorganisms. With the invasion of the rhizoid system, a series of CAZymes are secreted to release cellulose, hemicellulose, and oligosaccharides from lignocellulose and convert these polymers into soluble sugars. These sugars could be used by hosts and microbiota within the system
[29][17][38][39]. The important developmental stages of anaerobic fungi and their physical digestion of feedstuff can be clearly observed under a microscope (
Figure 1).
Figure 1. Anaerobic fungus Pecoramyces sp. F1 under the phase contrast microscope. The anaerobic fungus Pecoramyces sp. F1 undergoes the growth stages of zoospore attachment (A), rhizoid growth and penetration into the rice straw surface (B–F), and sporangium development and maturation (C–E).
2.2. Digestion by Diverse Plant Fiber-Degrading Enzymes
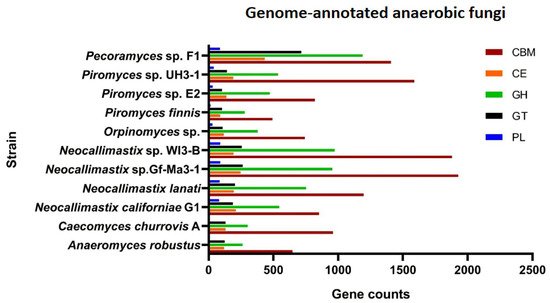
The efficient degradation of forage by anaerobic fungi is also attributed to the complete variety of carbohydrate-active enzymes (CAZymes), including glycoside hydrolases (GHs), carbohydrate esterases (CEs), polysaccharide lyases (PLs), glycosyl transferases (GTs). Carbohydrate-binding modules (CBMs) and less studied auxiliary activities (AAs) are accessories that are functionally related to CAZymes. Genome information of some anaerobic fungal strains that have been genetically annotated showed the presence of rich CAZymes, CBMs, and AAs of different types (
Figure 2). Details on activities of these enzymes can be found in another article
[35]. The most diverse group of CAZymes in the anaerobic fungi are the GHs
[35]. Enzymes hydrolyzing cellulose, hemicellulose, pectin, and other plant wall polysaccharides constitute a large group of GHs. Among these enzymes, cellulases and hemicellulases are the main ones.
Figure 2. Genome-annotated information of anaerobic fungi. Anaerobic fungi strains that have been genome-annotated (
Caecomyces churrovis A,
Anaeromyces robustus,
Neocallimastix californiae G1,
Neocallimastix lanati,
Neocallimastix sp. Gf-Ma3-1,
Neocallimastix sp. WI3-B,
Orpinomyces sp.,
Piromyces finnis,
Piromyces sp. E2,
Piromyces sp. UH3-1) can be found in JGI’s MycoCosm (
https://mycocosm.jgi.doe.gov/mycocosm/annotations/browser/cazy/summary;pEimlQ?p=neocallimastigomycetes (accessed on 10 December 2021)), and the results of their annotation in the CAZymes database are included. The genome of
Pecoramyces sp. F1 was uploaded to dbCAN2
[40] for online annotation, and at the same time, BLAST
[41] was used for the annotation of gene models against CAZymes database too. The data of AAs presented in the annotation results of
Pecoramyces sp. F1 but that of other 10 strains cannot be found in JGI’s MycoCosm (
https://mycocosm.jgi.doe.gov/mycocosm/annotations/browser/cazy/summary;pEimlQ?p=neocallimastigomycetes (accessed on 10 December 2021)), so the data of AAs are not shown. dbCAN2 and BLAST results were combined to get the
Pecoramyces sp. F1 genome annotation results in the figure.
Cellulase catalyzes the hydrolysis of β-1, 4-glucosidase in cellulose. Three types of cellulases (endoglucanase, exoglucanase, and β-glucosidase) that have been reported in anaerobic fungi together hydrolyze cellulose and release glucose
[42]. Endoglucanase is the most important component of the cellulase system, which can cleave the glucan chain inside the cellulose. High levels of endoglucanase were found in the culture supernatants of
Neocallimastix frontalis[43][44],
Piromyces communis[45], and
Orpinomyces sp.
[46]. Endoglucanase production was maximum when the fungi were grown on cellulose, whereas synthesis was totally repressed by the addition of glucose, indicating that the enzyme was subject to regulation
[43]. Exoglucanase cleaves cellobiose units, the building blocks of cellulose, from the ends of the cellulose chain. Exoglucanase active against microcrystalline cellulose was also detected, but at lower levels than endoglucanase
[43][46]. In addition, β-glucosidase cleaves cellobiose, which is a potent inhibitor of the former two enzymes, to yield glucose
[47]. Previous studies have classified that the glycoside hydrolase enzyme classes GH1, GH2, GH3, GH5, GH6, GH8, GH9, GH38, GH45, GH48, and GH74 encode for cellulases
[48]. Enzymes in different GHs have a different fiber hydrolysis activity according to their structure. The expression of these classes of enzymes varies according to fungal species and provided growth substrates. GH6 and GH5 were the two highest-expressed families in the
Pecoramyces sp. F1 according to its transcriptome analysis, whereas GH1 and GH33 were the lowest-expressed ones
[49]. In the genome of
Pecoramyces ruminantium C1A, GH45 and GH48 were highly expressed with glucose as substrate, while the expression of GH6 was dramatically improved by corn stover
[50].
Hemicellulose is the main component of the plant primary wall. It is a heteropolysaccharide containing xylan, glucuronoxylan, arabinoxylan, glucomannan, galactomannan, and xyloglucan
[51][52]. The component monosaccharides are connected to each other, forming a hard part of the cell wall to prevent microorganisms from using the plant cell contents. The hemicellulases secreted by anaerobic fungi include xylanases, mannanases, galactanases, β-glucanases and so on, which can degrade hemicellulose into various oligomers and monosaccharides. Due to xylan being the basic polymeric compound of hemicellulose and the high hydrolase activity of xylanases, xylanases are the most studied enzymes to date among the hemicellulose hydrolases from anaerobic fungi
[53][54][55]. Heterologous expression of these enzymes has also been developed. Xue et al. isolated a
Neocallimastix patricianun xylanase cDNA and engineered it for heterologous expression in
Escherichia coli (
E. coli). The modified xylanase produced in
E. colihad a specific activity of 1229 U mg
−1 protein at pH 7 and 50 °C, without purification
[56]. The genes encoding xylanases from
Neocallimastix sp. GMLF2
[57],
Orpinomyces sp. Strain 2, and
Neocallimastixfrontalis[58] were also cloned into
E. coli, and a high expression was obtained. In addition to
E. coli,
Hypocrea sp.
[59],
Kluyveromyces sp., and
Pichia sp.
[60] may also be ideal heterologous expression vectors for xylanases from anaerobic fungi. Successful development of heterologous production technologies for the enzymes from anaerobic fungi constitutes a significant advancement in applying this promising source of genes towards lignocellulose bioconversion. The glycoside hydrolase classes GH10, GH11, GH30, GH31, GH38, GH39, GH43, GH47, GH53, and GH115 encode for hemicellulases, respectively
[48]. Xylanases are representative enzymes of GH10 and GH11. The former has a relatively high molecular weight, while the latter has a lower molecular weight.
In addition to the free enzymes secreted outside the cell, anaerobic fungi also secrete a number of large (MDa) multiprotein cellulolytic complexes, known as cellulosomes, which can degrade crude fibers more completely and efficiently. Although most studies have reported the function of cellulosomes in anaerobic microorganisms, such as anaerobic fungi, and the bacteria
Clostridium thermocellum and
Ruminococcus albus, there have also been reports on the discovery of cellulosomes in aerobic bacteria
[61]. Cellulosomes include carbohydrate-binding domains, noncatalytic protein domains, and many glycosyl-hydrolases (cellulases, hemicellulases, pectin enzymes, chitinases, and other enzymes). The assembly of cellulosomes involves interactions between dockerin domains on different catalytic enzyme subunits and cohesin domain on noncatalytic scaffolding, and the specific interactions between them allow various enzyme proteins to bind stably in this supramolecular structure. Scaffold proteins and some enzymes contain CBMs, which promote the binding of the cellulosome enzyme to substrate cellulose. Cellulosomes are attached to the cell surface by additional anchoring domains by noncovalent bonding
[62]. Many GHs of anaerobic fungi are involved in the formation of cellulosomes. It was reported that anaerobic fungal cellulosomes exhibit additional 13% higher GH activity due to the presence of GH3, GH6, and GH45 compared with bacterial cellulosomes; especially the supplementary β-glucosidase conferred by GH3 activity empowers fungal cellulosomes in converting cellulose to single simple sugars (monosaccharides) when compared with low-molecular-weight oligosaccharide-generating bacterial cellulosomes
[30][63]. Because of the powerful fiber degradation capability of cellulosomes, better than commercial preparations containing noncomplexed enzymes, the efficient degradation of cheap fibrous materials is possible if the modified cellulosome genes can be inserted into appropriate host cells and expressed using genetic engineering