Neurological/neurovascular disorders constitute the leading cause of disability and the second leading cause of death globally. Major neurological/neurovascular disorders or diseases include cerebral stroke, Alzheimer’s disease, spinal cord injury, neonatal hypoxic-ischemic encephalopathy, and others. Their pathophysiology is considered highly complex and is the main obstacle in developing any drugs for these diseases. Studies have demonstrated the highly promising effects of sovateltide on the neurovascular system and improved recovery, comprehensively after various injuries/insults, which have generated immense hope of developing an effective therapy with the potential to treat various neurological disorders.
- endothelin B receptors
- neurological diseases
- neurovascular disorders
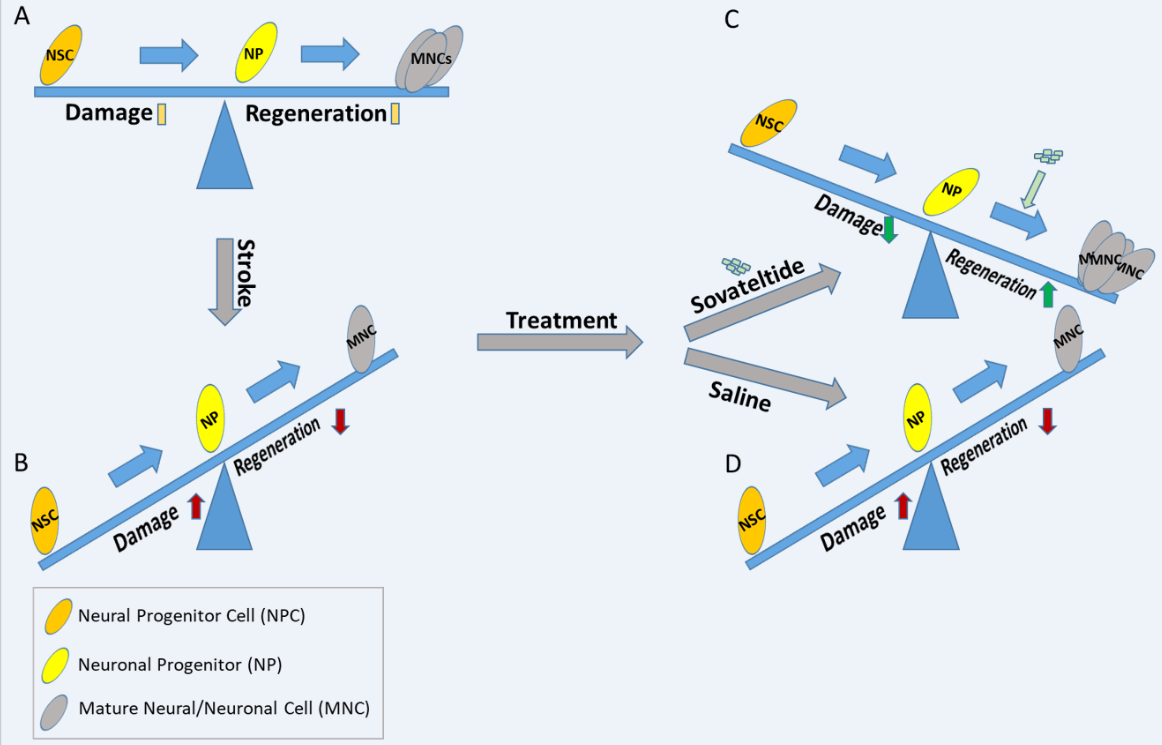
- sovateltide
- IRL-1620
- neurogenesis
- stem/progenitor cells
- regeneration
1. Stroke
2. Alzheimer’s Disease
3. Spinal Cord Injury
4. Neonatal Hypoxic-Ischemic Encephalopathy
This entry is adapted from the peer-reviewed paper 10.3390/ijms23063146
References
- Donkor, E.S. Stroke in the 21(st) Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat. 2018, 2018, 3238165.
- Bamford, J.; Sandercock, P.; Dennis, M.; Burn, J.; Warlow, C. Classification and natural history of clinically identifiable subtypes of cerebral infarction. Lancet 1991, 337, 1521–1526.
- Seet, R.C.S.; Rabinstein, A.A. Symptomatic Intracranial Hemorrhage following Intravenous Thrombolysis for Acute Ischemic Stroke: A Critical Review of Case Definitions. Cerebrovasc. Dis. 2012, 34, 106–114.
- Minnerup, J.; Wersching, H.; Schilling, M.; Schabitz, W.R. Analysis of early phase and subsequent phase III stroke studies of neuroprotectants: Outcomes and predictors for success. Exp. Transl. Stroke Med. 2014, 6, 2.
- Gulati, A.; Hornick, M.G.; Briyal, S.; Lavhale, M.S. A novel neuroregenerative approach using ET(B) receptor agonist, IRL-1620, to treat CNS disorders. Physiol. Res. 2018, 67, S95–S113.
- Lampl, Y.; Fleminger, G.; Gilad, R.; Galron, R.; Sarova-Pinhas, I.; Sokolovsky, M. Endothelin in cerebrospinal fluid and plasma of patients in the early stage of ischemic stroke. Stroke J. Cereb. Circ. 1997, 28, 1951–1955.
- Ziv, I.; Fleminger, G.; Djaldetti, R.; Achiron, A.; Melamed, E.; Sokolovsky, M. Increased plasma endothelin-1 in acute ischemic stroke. Stroke J. Cereb. Circ. 1992, 23, 1014–1016.
- Barone, F.C.; Ohlstein, E.H.; Hunter, A.J.; Campbell, C.A.; Hadingham, S.H.; Parsons, A.A.; Yang, Y.; Shohami, E. Selective antagonism of endothelin-A-receptors improves outcome in both head trauma and focal stroke in rat. J. Cardiovasc. Pharmacol. 2000, 36, S357–S361.
- Briyal, S.; Gulati, A. Endothelin-A receptor antagonist BQ123 potentiates acetaminophen induced hypothermia and reduces infarction following focal cerebral ischemia in rats. Eur. J. Pharmacol. 2010, 644, 73–79.
- Legos, J.J.; Lenhard, S.C.; Haimbach, R.E.; Schaeffer, T.R.; Bentley, R.G.; McVey, M.J.; Chandra, S.; Irving, E.A.; Andrew, A.P.; Barone, F.C. SB 234551 selective ET(A) receptor antagonism: Perfusion/diffusion MRI used to define treatable stroke model, time to treatment and mechanism of protection. Exp. Neurol. 2008, 212, 53–62.
- Tatlisumak, T.; Carano, R.A.; Takano, K.; Opgenorth, T.J.; Sotak, C.H.; Fisher, M. A novel endothelin antagonist, A-127722, attenuates ischemic lesion size in rats with temporary middle cerebral artery occlusion: A diffusion and perfusion MRI study. Stroke J. Cereb. Circ. 1998, 29, 850–857; discussion 857–858.
- Zhang, R.L.; Zhang, C.; Zhang, L.; Roberts, C.; Lu, M.; Kapke, A.; Cui, Y.; Ninomiya, M.; Nagafuji, T.; Albala, B.; et al. Synergistic effect of an endothelin type A receptor antagonist, S-0139, with rtPA on the neuroprotection after embolic stroke. Stroke J. Cereb. Circ. 2008, 39, 2830–2836.
- Briyal, S.; Gulati, A.; Gupta, Y.K. Effect of combination of endothelin receptor antagonist (TAK-044) and aspirin in middle cerebral artery occlusion model of acute ischemic stroke in rats. Methods Find. Exp. Clin. Pharmacol. 2007, 29, 257–263.
- Briyal, S.; Pant, A.B.; Gupta, Y.K. Protective effect of endothelin antagonist (TAK-044) on neuronal cell viability in in vitro oxygen-glucose deprivation model of stroke. Indian J. Physiol. Pharmacol. 2006, 50, 157–162.
- Chuquet, J.; Benchenane, K.; Toutain, J.; MacKenzie, E.T.; Roussel, S.; Touzani, O. Selective blockade of endothelin-B receptors exacerbates ischemic brain damage in the rat. Stroke J. Cereb. Circ. 2002, 33, 3019–3025.
- Ehrenreich, H.; Oldenburg, J.; Hasselblatt, M.; Herms, J.; Dembowski, C.; Loffler, B.M.; Bruck, W.; Kamrowski-Kruck, H.; Gall, S.; Siren, A.L.; et al. Endothelin B receptor-deficient rats as a subtraction model to study the cerebral endothelin system. Neuroscience 1999, 91, 1067–1075.
- Ehrenreich, H.; Nau, T.R.; Dembowski, C.; Hasselblatt, M.; Barth, M.; Hahn, A.; Schilling, L.; Siren, A.L.; Bruck, W. Endothelin b receptor deficiency is associated with an increased rate of neuronal apoptosis in the dentate gyrus. Neuroscience 2000, 95, 993–1001.
- Castaneda, M.M.; Cubilla, M.A.; Lopez-Vicchi, M.M.; Suburo, A.M. Endothelinergic cells in the subependymal region of mice. Brain Res. 2010, 1321, 20–30.
- Gulati, A.; Kumar, A.; Morrison, S.; Shahani, B.T. Effect of centrally administered endothelin agonists on systemic and regional blood circulation in the rat: Role of sympathetic nervous system. Neuropeptides 1997, 31, 301–309.
- Koyama, Y.; Takemura, M.; Fujiki, K.; Ishikawa, N.; Shigenaga, Y.; Baba, A. BQ788, an endothelin ET(B) receptor antagonist, attenuates stab wound injury-induced reactive astrocytes in rat brain. Glia 1999, 26, 268–271.
- Sakurai-Yamashita, Y.; Niwa, M.; Yamashita, K.; Kataoka, Y.; Himeno, A.; Shigematsu, K.; Tsutsumi, K.; Taniyama, K. Endothelin receptors in kainic acid-induced neural lesions of rat brain. Neuroscience 1997, 81, 565–577.
- Yamashita, K.; Niwa, M.; Kataoka, Y.; Shigematsu, K.; Himeno, A.; Tsutsumi, K.; Nakano-Nakashima, M.; Sakurai-Yamashita, Y.; Shibata, S.; Taniyama, K. Microglia with an endothelin ETB receptor aggregate in rat hippocampus CA1 subfields following transient forebrain ischemia. J. Neurochem. 1994, 63, 1042–1051.
- Vidovic, M.; Chen, M.M.; Lu, Q.Y.; Kalloniatis, K.F.; Martin, B.M.; Tan, A.H.; Lynch, C.; Croaker, G.D.; Cass, D.T.; Song, Z.M. Deficiency in endothelin receptor B reduces proliferation of neuronal progenitors and increases apoptosis in postnatal rat cerebellum. Cell. Mol. Neurobiol. 2008, 28, 1129–1138.
- Siren, A.L.; Lewczuk, P.; Hasselblatt, M.; Dembowski, C.; Schilling, L.; Ehrenreich, H. Endothelin B receptor deficiency augments neuronal damage upon exposure to hypoxia-ischemia in vivo. Brain Res. 2002, 945, 144–149.
- Leonard, M.G.; Prazad, P.; Puppala, B.; Gulati, A. Selective Endothelin-B Receptor Stimulation Increases Vascular Endothelial Growth Factor in the Rat Brain during Postnatal Development. Drug Res. 2015, 65, 607–613.
- Puppala, B.; Awan, I.; Briyal, S.; Mbachu, O.; Leonard, M.; Gulati, A. Ontogeny of endothelin receptors in the brain, heart, and kidneys of neonatal rats. Brain Dev. 2015, 37, 206–215.
- Gulati, A. Endothelin Receptors, Mitochondria and Neurogenesis in Cerebral Ischemia. Curr. Neuropharmacol. 2016, 14, 619–626.
- Leonard, M.G.; Briyal, S.; Gulati, A. Endothelin B receptor agonist, IRL-1620, reduces neurological damage following permanent middle cerebral artery occlusion in rats. Brain Res. 2011, 1420, 48–58.
- Leonard, M.G.; Briyal, S.; Gulati, A. Endothelin B receptor agonist, IRL-1620, provides long-term neuroprotection in cerebral ischemia in rats. Brain Res. 2012, 1464, 14–23.
- Leonard, M.G.; Gulati, A. Endothelin B receptor agonist, IRL-1620, enhances angiogenesis and neurogenesis following cerebral ischemia in rats. Brain Res. 2013, 1528, 28–41.
- Briyal, S.; Ranjan, A.K.; Hornick, M.G.; Puppala, A.K.; Luu, T.; Gulati, A. Anti-apoptotic activity of ETB receptor agonist, IRL-1620, protects neural cells in rats with cerebral ischemia. Sci. Rep. 2019, 9, 10439.
- Cifuentes, E.G.; Hornick, M.G.; Havalad, S.; Donovan, R.L.; Gulati, A. Neuroprotective Effect of IRL-1620, an Endothelin B Receptor Agonist, on a Pediatric Rat Model of Middle Cerebral Artery Occlusion. Front. Pediatr. 2018, 6, 310.
- Ranjan, A.K.; Briyal, S.; Gulati, A. Sovateltide (IRL-1620) activates neuronal differentiation and prevents mitochondrial dysfunction in adult mammalian brains following stroke. Sci. Rep. 2020, 10, 12737.
- Ranjan, A.K.; Briyal, S.; Khandekar, D.; Gulati, A. Sovateltide (IRL-1620) affects neuronal progenitors and prevents cerebral tissue damage after ischemic stroke. Can. J. Physiol. Pharmacol. 2020, 98, 659–666.
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32.
- 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020.
- Crous-Bou, M.; Minguillon, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer’s disease prevention: From risk factors to early intervention. Alzheimer’s Res. Ther. 2017, 9, 71.
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res 2018, 7, 1161.
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031.
- Nisbet, R.M.; Polanco, J.C.; Ittner, L.M.; Gotz, J. Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol. 2015, 129, 207–220.
- Cummings, J.L.; Ringman, J.; Vinters, H.V. Neuropathologic correlates of trial-related instruments for Alzheimer’s disease. Am. J. Neurodegener. Dis. 2014, 3, 45–49.
- Emanuel, E.J. A Middle Ground for Accelerated Drug Approval-Lessons from Aducanumab. JAMA 2021, 326, 1367–1368.
- Dunn, B.; Stein, P.; Temple, R.; Cavazzoni, P. An Appropriate Use of Accelerated Approval—Aducanumab for Alzheimer’s Disease. N. Engl. J. Med. 2021, 385, 856–857.
- Alexander, G.C.; Knopman, D.S.; Emerson, S.S.; Ovbiagele, B.; Kryscio, R.J.; Perlmutter, J.S.; Kesselheim, A.S. Revisiting FDA Approval of Aducanumab. N. Engl. J. Med. 2021, 385, 769–771.
- Gulati, A.; Agrawal, N.; Vibha, D.; Misra, U.K.; Paul, B.; Jain, D.; Pandian, J.; Borgohain, R. Safety and Efficacy of Sovateltide (IRL-1620) in a Multicenter Randomized Controlled Clinical Trial in Patients with Acute Cerebral Ischemic Stroke. CNS Drugs 2021, 35, 85–104.
- Palmer, J.C.; Baig, S.; Kehoe, P.G.; Love, S. Endothelin-converting enzyme-2 is increased in Alzheimer’s disease and up-regulated by Abeta. Am. J. Pathol. 2009, 175, 262–270.
- Koyama, Y. Endothelin systems in the brain: Involvement in pathophysiological responses of damaged nerve tissues. Biomol. Concepts 2013, 4, 335–347.
- Turner, A.J.; Murphy, L.J. Molecular pharmacology of endothelin converting enzymes. Biochem. Pharmacol. 1996, 51, 91–102.
- Rodriguiz, R.M.; Gadnidze, K.; Ragnauth, A.; Dorr, N.; Yanagisawa, M.; Wetsel, W.C.; Devi, L.A. Animals lacking endothelin-converting enzyme-2 are deficient in learning and memory. Genes Brain Behav. 2008, 7, 418–426.
- Eckman, E.A.; Watson, M.; Marlow, L.; Sambamurti, K.; Eckman, C.B. Alzheimer’s disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J. Biol. Chem. 2003, 278, 2081–2084.
- Yamashita, K.I.; Taniwaki, Y.; Utsunomiya, H.; Taniwaki, T. Cerebral blood flow reduction associated with orientation for time in amnesic mild cognitive impairment and Alzheimer disease patients. J. Neuroimaging 2014, 24, 590–594.
- Gulati, A.; Kumar, A.; Shahani, B.T. Cardiovascular effects of centrally administered endothelin-1 and its relationship to changes in cerebral blood flow. Life Sci. 1996, 58, 437–445.
- Gulati, A.; Rebello, S.; Roy, S.; Saxena, P.R. Cardiovascular effects of centrally administered endothelin-1 in rats. J. Cardiovasc. Pharmacol. 1995, 26 (Suppl. 3), S244–S246.
- Paris, D.; Humphrey, J.; Quadros, A.; Patel, N.; Crescentini, R.; Crawford, F.; Mullan, M. Vasoactive effects of A beta in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer’s disease: Role of inflammation. Neurol. Res. 2003, 25, 642–651.
- Briyal, S.; Philip, T.; Gulati, A. Endothelin-A receptor antagonists prevent amyloid-beta-induced increase in ETA receptor expression, oxidative stress, and cognitive impairment. J. Alzheimer’s Dis. 2011, 23, 491–503.
- Druckenbrod, N.R.; Powers, P.A.; Bartley, C.R.; Walker, J.W.; Epstein, M.L. Targeting of endothelin receptor-B to the neural crest. Genesis 2008, 46, 396–400.
- Nishikawa, K.; Ayukawa, K.; Hara, Y.; Wada, K.; Aoki, S. Endothelin/endothelin-B receptor signals regulate ventricle-directed interkinetic nuclear migration of cerebral cortical neural progenitors. Neurochem. Int. 2011, 58, 261–272.
- Yagami, T.; Ueda, K.; Asakura, K.; Kuroda, T.; Hata, S.; Sakaeda, T.; Kambayashi, Y.; Fujimoto, M. Effects of endothelin B receptor agonists on amyloid beta protein (25–35)-induced neuronal cell death. Brain Res. 2002, 948, 72–81.
- Leonard, M.G.; Gulati, A. Repeated administration of ET(B) receptor agonist, IRL-1620, produces tachyphylaxis only to its hypotensive effect. Pharmacol. Res. 2009, 60, 402–410.
- Joshi, M.D.; Oesterling, B.M.; Wu, C.; Gwizdz, N.; Pais, G.; Briyal, S.; Gulati, A. Evaluation of liposomal nanocarriers loaded with ETB receptor agonist, IRL-1620, using cell-based assays. Neuroscience 2016, 312, 141–152.
- Briyal, S.; Shepard, C.; Gulati, A. Endothelin receptor type B agonist, IRL-1620, prevents beta amyloid (Abeta) induced oxidative stress and cognitive impairment in normal and diabetic rats. Pharmacol. Biochem. Behav. 2014, 120, 65–72.
- Briyal, S.; Nguyen, C.; Leonard, M.; Gulati, A. Stimulation of endothelin B receptors by IRL-1620 decreases the progression of Alzheimer’s disease. Neuroscience 2015, 301, 1–11.
- Alizadeh, A.; Dyck, S.M.; Karimi-Abdolrezaee, S. Traumatic Spinal Cord Injury: An Overview of Pathophysiology, Models and Acute Injury Mechanisms. Front. Neurol. 2019, 10, 282.
- Nori, S.; Ahuja, C.S.; Fehlings, M.G. Translational Advances in the Management of Acute Spinal Cord Injury: What is New? What is Hot? Neurosurgery 2017, 64, 119–128.
- Donovan, J.; Kirshblum, S. Clinical Trials in Traumatic Spinal Cord Injury. Neurotherapeutics 2018, 15, 654–668.
- Peters, C.M.; Rogers, S.D.; Pomonis, J.D.; Egnaczyk, G.F.; Keyser, C.P.; Schmidt, J.A.; Ghilardi, J.R.; Maggio, J.E.; Mantyh, P.W. Endothelin receptor expression in the normal and injured spinal cord: Potential involvement in injury-induced ischemia and gliosis. Exp. Neurol. 2003, 180, 1–13.
- Kallakuri, S.; Kreipke, C.W.; Schafer, P.C.; Schafer, S.M.; Rafols, J.A. Brain cellular localization of endothelin receptors A and B in a rodent model of diffuse traumatic brain injury. Neuroscience 2010, 168, 820–830.
- McKenzie, A.L.; Hall, J.J.; Aihara, N.; Fukuda, K.; Noble, L.J. Immunolocalization of endothelin in the traumatized spinal cord: Relationship to blood-spinal cord barrier breakdown. J. Neurotrauma 1995, 12, 257–268.
- Guo, J.; Li, Y.; He, Z.; Zhang, B.; Li, Y.; Hu, J.; Han, M.; Xu, Y.; Li, Y.; Gu, J.; et al. Targeting endothelin receptors A and B attenuates the inflammatory response and improves locomotor function following spinal cord injury in mice. Int. J. Mol. Med. 2014, 34, 74–82.
- Forner, S.; Martini, A.C.; Andrade, E.L.; Rae, G.A. Neuropathic pain induced by spinal cord injury: Role of endothelin ETA and ETB receptors. Neurosci. Lett. 2016, 617, 14–21.
- Uesugi, M.; Kasuya, Y.; Hayashi, K.; Goto, K. SB209670, a potent endothelin receptor antagonist, prevents or delays axonal degeneration after spinal cord injury. Brain Res. 1998, 786, 235–239.
- Laziz, I.; Larbi, A.; Grebert, D.; Sautel, M.; Congar, P.; Lacroix, M.C.; Salesse, R.; Meunier, N. Endothelin as a neuroprotective factor in the olfactory epithelium. Neuroscience 2011, 172, 20–29.
- Yagami, T.; Ueda, K.; Sakaeda, T.; Okamura, N.; Nakazato, H.; Kuroda, T.; Hata, S.; Sakaguchi, G.; Itoh, N.; Hashimoto, Y.; et al. Effects of an endothelin B receptor agonist on secretory phospholipase A2-IIA-induced apoptosis in cortical neurons. Neuropharmacology 2005, 48, 291–300.
- Kurinczuk, J.J.; White-Koning, M.; Badawi, N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum. Dev. 2010, 86, 329–338.
- Lawn, J.E.; Blencowe, H.; Oza, S.; You, D.; Lee, A.C.; Waiswa, P.; Lalli, M.; Bhutta, Z.; Barros, A.J.; Christian, P.; et al. Every Newborn: Progress, priorities, and potential beyond survival. Lancet 2014, 384, 189–205.
- Liu, L.; Oza, S.; Hogan, D.; Chu, Y.; Perin, J.; Zhu, J.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of under-5 mortality in 2000-15: An updated systematic analysis with implications for the Sustainable Development Goals. Lancet 2016, 388, 3027–3035.
- Harteman, J.C.; Nikkels, P.G.; Benders, M.J.; Kwee, A.; Groenendaal, F.; de Vries, L.S. Placental pathology in full-term infants with hypoxic-ischemic neonatal encephalopathy and association with magnetic resonance imaging pattern of brain injury. J. Pediatr. 2013, 163, 968–995.e2.
- Azzopardi, D.; Wyatt, J.S.; Cady, E.B.; Delpy, D.T.; Baudin, J.; Stewart, A.L.; Hope, P.L.; Hamilton, P.A.; Reynolds, E.O. Prognosis of newborn infants with hypoxic-ischemic brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatr. Res. 1989, 25, 445–451.
- Azzopardi, D.V.; Strohm, B.; Edwards, A.D.; Dyet, L.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; Levene, M.; Marlow, N.; Porter, E.; et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N. Engl. J. Med. 2009, 361, 1349–1358.
- Bale, G.; Mitra, S.; de Roever, I.; Sokolska, M.; Price, D.; Bainbridge, A.; Gunny, R.; Uria-Avellanal, C.; Kendall, G.S.; Meek, J.; et al. Oxygen dependency of mitochondrial metabolism indicates outcome of newborn brain injury. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2019, 39, 2035–2047.
- Yildiz, E.P.; Ekici, B.; Tatli, B. Neonatal hypoxic ischemic encephalopathy: An update on disease pathogenesis and treatment. Expert Rev. Neurother. 2017, 17, 449–459.
- Jacobs, S.E.; Morley, C.J.; Inder, T.E.; Stewart, M.J.; Smith, K.R.; McNamara, P.J.; Wright, I.M.; Kirpalani, H.M.; Darlow, B.A.; Doyle, L.W.; et al. Whole-body hypothermia for term and near-term newborns with hypoxic-ischemic encephalopathy: A randomized controlled trial. Arch. Pediatr. Adolesc. Med. 2011, 165, 692–700.
- Gluckman, P.D.; Wyatt, J.S.; Azzopardi, D.; Ballard, R.; Edwards, A.D.; Ferriero, D.M.; Polin, R.A.; Robertson, C.M.; Thoresen, M.; Whitelaw, A.; et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: Multicentre randomised trial. Lancet 2005, 365, 663–670.
- Shankaran, S.; Laptook, A.R.; Ehrenkranz, R.A.; Tyson, J.E.; McDonald, S.A.; Donovan, E.F.; Fanaroff, A.A.; Poole, W.K.; Wright, L.L.; Higgins, R.D.; et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N. Engl. J. Med. 2005, 353, 1574–1584.
- Edwards, A.D.; Brocklehurst, P.; Gunn, A.J.; Halliday, H.; Juszczak, E.; Levene, M.; Strohm, B.; Thoresen, M.; Whitelaw, A.; Azzopardi, D. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: Synthesis and meta-analysis of trial data. BMJ 2010, 340, c363.
- Shankaran, S.; Pappas, A.; McDonald, S.A.; Vohr, B.R.; Hintz, S.R.; Yolton, K.; Gustafson, K.E.; Leach, T.M.; Green, C.; Bara, R.; et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N. Engl. J. Med. 2012, 366, 2085–2092.
- Gunn, A.J.; Gluckman, P.D.; Gunn, T.R. Selective head cooling in newborn infants after perinatal asphyxia: A safety study. Pediatrics 1998, 102, 885–892.
- Higgins, R.D.; Raju, T.; Edwards, A.D.; Azzopardi, D.V.; Bose, C.L.; Clark, R.H.; Ferriero, D.M.; Guillet, R.; Gunn, A.J.; Hagberg, H.; et al. Hypothermia and other treatment options for neonatal encephalopathy: An executive summary of the Eunice Kennedy Shriver NICHD workshop. J. Pediatr. 2011, 159, 851–858.e1.
- Thoresen, M.; Whitelaw, A. Cardiovascular changes during mild therapeutic hypothermia and rewarming in infants with hypoxic-ischemic encephalopathy. Pediatrics 2000, 106, 92–99.