Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
|
Obstetrics & Gynaecology
Paclitaxel (PTX), the most widely used anticancer drug, is applied for the treatment of various types of malignant diseases. Mechanisms of PTX action represent several ways in which PTX affects cellular processes resulting in programmed cell death. PTX is frequently used as the first-line treatment drug in breast cancer (BC).
- breast cancer
- anti-cancer therapy
- chemotherapy
- Paclitaxel
1. Introduction
A crucial aspect of the modern era is the rapid progress in the prevalence of many civilization diseases including cancer [1]. Breast cancer (BC )is the most commonly occurring malignant disease in women and the leading cause of cancer death among them and still remains a global problem of public health [2]. Modern approaches in the field of oncology aimed at BC including novelties in diagnosis, treatment, and prevention have a crucial role in the management of cancer. Better knowledge of the biologic heterogeneity of BC leads to the development of more effective therapy concepts in personalized medicine [3]. Over the last decades, substantial progress in the treatment of BC led to the discovery of new drugs with specific actions in cancer suppression. Currently, there are several classes of chemo-therapeuticals based on antimetabolites, alkylating agents, immunological elements, hormonal components, or mitotic deprivation [4]. Recently, two groups of chemotherapeutic drugs (anthracyclines and taxanes) were widely used in adjuvant and neoadjuvant treatment of BC [5]. Paclitaxel (PTX), a class of taxanes, is an antineoplastic drug with an impact on the stabilization of microtubules, which represents a widely used chemotherapeutic agent in numerous cancers. The effect of PTX as an antimitotic drug was documented in a large number of studies [6][7][8]. Moreover, the mechanisms of PTX action associated with the inhibition of tumor growth can act on different levels. In these studies, PTX initiated a cascade of signaling pathways resulting in programmed cell death [9][10]. Modulation of epigenetic markers represents the novelty of cancer-related research, and PTX may also regulate the expression of certain microRNAs (miRNAs) associated with cancer progression. Furthermore, PTX can exert a variety of positive influences on the modulation of immune response via regulation of chemokines, cytokines, or immune cells [11][12]. The resistance of BC to PTX and other chemotherapeutics as a consequence of disequilibrium in various signaling pathways, mutations in certain genes, and epigenetic deregulations is responsible for the worse clinical outcome for patients with BC [13][14][15]. The global challenge in the application of PTX as a dominant anticancer chemotherapeutic agent is the reduction of side effects and increasing drug efficiency. Novelties such as Albumin-bound PTX(nab-PTX) are awesome examples of the progress in the oncology-associated area focused on cross-connection of nanotechnology and cancer treatment [16].
2. Paclitaxel: Fundamental Drug in Chemotherapy and Novel Advances in its Application
As noted above, antimitotic chemotherapeutics suppress the polymerization dynamic of microtubules, resulting in the induction of mitotic arrests as a consequence of the activation of the mitotic checkpoint. Consequently, PTX, a member of taxanes, represents one of the most important antineoplastic drugs frequently used in the treatment of numerous types of cancers including BC.
2.1. The Origin of Paclitaxel
PTX, trademarked by Bristol-Myers Squibb (BMS) as Taxol™ in 1992 [17], is an antimitotic, anticancer drug that was approved by the FDA (Food and Drug Administration) in 1994 for use in BC [18]. PTX is clinically used to treat solid tumors such as ovarian cancer, hormone refractory prostate cancer, and non-small cell lung cancer [19]. The active ingredient was first isolated from the Pacific Yew tree (Taxus brevifolia) by Mansukh Wani and Monroe Wall [20]. The drug went through additional clinical trials and testing on mouse tumor models before it was FDA approved for ovarian cancer in 1992 [17]. Due to the high demand for the drug, the slow-growing T. brevifolia tree was not able to provide the market and research’s needs; therefore, it was concluded that the production of PTX from T. brevifolia was impractical, non-environmentally conscious, and financially burdening according to the National Cancer Institute (NCI) [21][22]. In 1993, a new method of mass-producing PTX was associated with a fungus isolated from the phloem of T. brevifolia [23]. In 1994, a successful semi-synthetic approach of synthesizing PTX was formulated and approved by the FDA, which is the method of production until today [18][22].
2.2. Paclitaxel’s Mechanism of Action
(I) Paclitaxel as a Polymerization Factor
PTX binds to microtubules instead of tubulin dimers and stabilizes microtubules (polymerization) by promoting the assembly of alpha and beta tubulin subunits, the building blocks of microtubules [17][24]. The drug reduces the critical concentration of tubulin required for its assembly, therefore promoting the lengthening of the tubulin polymer [25]. The stability of the microtubules interferes with microtubules´ dynamics. Subsequently, the cell’s ability to divide is disrupted due to insufficient requirements of the mitotic checkpoint; therefore, cell division halts at the G2 or M phase. The polymerized and stable microtubules remain largely unaffected even by cold temperatures and calcium. The presence of calcium reduces PTX’s affinity for tubulin; therefore, the equilibrium of polymerization/depolymerization shifts towards polymerization to offset this effect [17][26][27]. Moreover, chondrocytes show that PTX causes cytoskeletal abnormalities in which microtubules become stubby and straight in the cytoplasm, with rough endoplasmic reticulum as opposed to fine, sinous filaments in the control group. These changes persist for 48 h after the removal of PTX. The changed microtubules dislodge ribosomes off the rough endoplasmic reticulum and fuse nearby endoplasmic reticulum complexes together [26]. PTX polymerizes only free microtubules not attached to or preexisting in the microtubule organizing centers (MTOC). Attached microtubules disappear in the presence of PTX [28]. PTX interferes with the dynamics of microtubules and microtubule polymerization and delays the progression of mitosis by inducing failure in chromosomal segregation, all of which eventually lead to the induction of apoptosis and mitotic arrest [29][30][31].
(II) Paclitaxel’s Effect Depends on Concentration
The mechanism of PTX cytotoxicity highly depends on the concentration of the drug in the cell as demonstrated in in vitro studies. Giannakakou et al. [32] documented that the reduction of proliferation of the lung carcinoma cell line A549 as well as breast MCF-7 cells after treatment by PTX at concentrations above 12 nM resulted in G2/M arrest. Interestingly, lower concentrations of PTX (3–6 nM) exerted similar potential to suppress the proliferation of cancer cells, resulting in programmed cell death [32]. A study focusing on drug concentration analyzed the effect of low doses of PTX (10 nM) on cancer cell invasiveness. In this in vitro study, researchers evaluated the impact of the non-anti-mitotic concentration of PTX resulting in the reduction of transwell invasion of MDA-MB-231 as a consequence of the regulation of voltage-dependent sodium channel expression [33]. Additionally, the potential role of low doses of PTX (20 nM) combined with a Wnt signaling inhibitor regulated the molecular events, including E-cadherin upregulation and β-catenin reduction, leading to suppression of tumor growth, metastasis, and angiogenesis in BC [34].
(III) Paclitaxel Affects Phosphorylation of Bcl-2
Although many researchers agree that the cytotoxicity of PTX lies in its ability to cause Bcl-2 hyperphosphorylation, many other studies report dephosphorylation of Bcl-2 coinciding with apoptosis [35][36][37]. However, since apoptosis does not occur immediately after exposure to PTX, the duration of exposure and constant Bcl-2 phosphorylation contribute to the drug’s cytotoxicity [38]. On the other hand, different studies proved that phosphorylated Bcl-2 does not dimerize with BAX; therefore, it is argued that the unassociated BAX favors apoptosis [39][40]. However, the later studies are older and probably less updated than the former studies, and these conclusions are based on prostate and leukemia cell lines.
(IV) Paclitaxel Affects Calcium Signaling
PTX induces the depletion of calcium ions from the mitochondrial reserve through the mitochondrial permeability transition pore (PTP). The leaving calcium induces PTP to release the apoptogenic factor cytochrome C (cyto C) into the cytosol from the mitochondria to initiate apoptosis [41][42][43]. It is believed that side effects of antimitotic drugs are related to the role of those drugs in the calcium signal cascade. The severity and heterogeneity of these side effects are attributed to the alterations in mitochondrial calcium uptake in different cells. Using extremely high doses of PTX results in the rupture of the mitochondria, the release of cyto C, and the initiation of apoptosis without the efflux of calcium [44]. Reasons why peripheral neurons are primarily affected by PTX are not known, even though most cells have mitochondria. On the other hand, PTX induces apoptosis in the presence of an extracellular calcium reservoir via calcium influx when the drug is administered in high doses. However, low doses of PTX demonstrated patterns of apoptosis independent of the extracellular concentration of calcium [10].
(V) The impact of Paclitaxel on microRNA Expression Profiles
MiRNA, small non-coding RNA with a regulatory function in gene expression, can be regulated by various antineoplastic drugs including PTX. Several studies focusing on miRNA expression demonstrated cross-connection between an application of the drug and alterations in miRNA expression profiles. After PTX intervention, expression levels of let-7a and miR-205 with tumor suppressor potential targeting K-Ras and HER3 were changed in the BC cell line BT-474 [45]. Interestingly, metronomic treatment (low dose LDM) by PTX reduced the level of let-7f, while the expression of thrombospondin-1 (TSP-1) associated with anti-angiogenic potential was increased in PTX LDM therapy [46]. In summary, preclinical trials demonstrated the modulatory potential of PTX in the regulation of miRNA expression, but further research is needed for a better implication of the drug in BC treatment.
(VI) The Immunomodulatory Effects of Paclitaxel
The role of PTX was also documented in the area of immunomodulation, with both stimulation and suppression of immune cells associated with tumor growth. On the other hand, the suppression of immune cells could have a negative impact on the host immune response against cancer development [47]. Increasing evidence supports participation of PTX in the regulation of host immunity via stimulation of macrophages leading to cytokine secretion including TNF-α or IL-12 that induce activation of natural killers (NK), dendric cells (DC), and cytotoxic T lymphocytes resulting in the eradication of tumor cells [11][48]. Additionally, direct-acting PTX was evaluated in DC via binding to Toll-like receptor localized on the DC surface, thus promoting maturation of antigen-presenting cells [49]. Furthermore, the dose-dependent administration of PTX led to an increased level of MHC class II [50]. The impact of PTX on immunomodulation was also identified in NK cells. A higher level of cytotoxicity correlated with an increased level of perforin representing the crucial effector protein of NK activity, resulting in the premise that PTX enhanced NK cytotoxicity in a dose-dependent manner [51]. Further research focusing on the regulation of the immune response in cancer is necessary for better understanding of an association between effects of PTX and immunopharmacology. The mechanisms of PTX in antineoplastic processes described above are summarized in Figure 1.
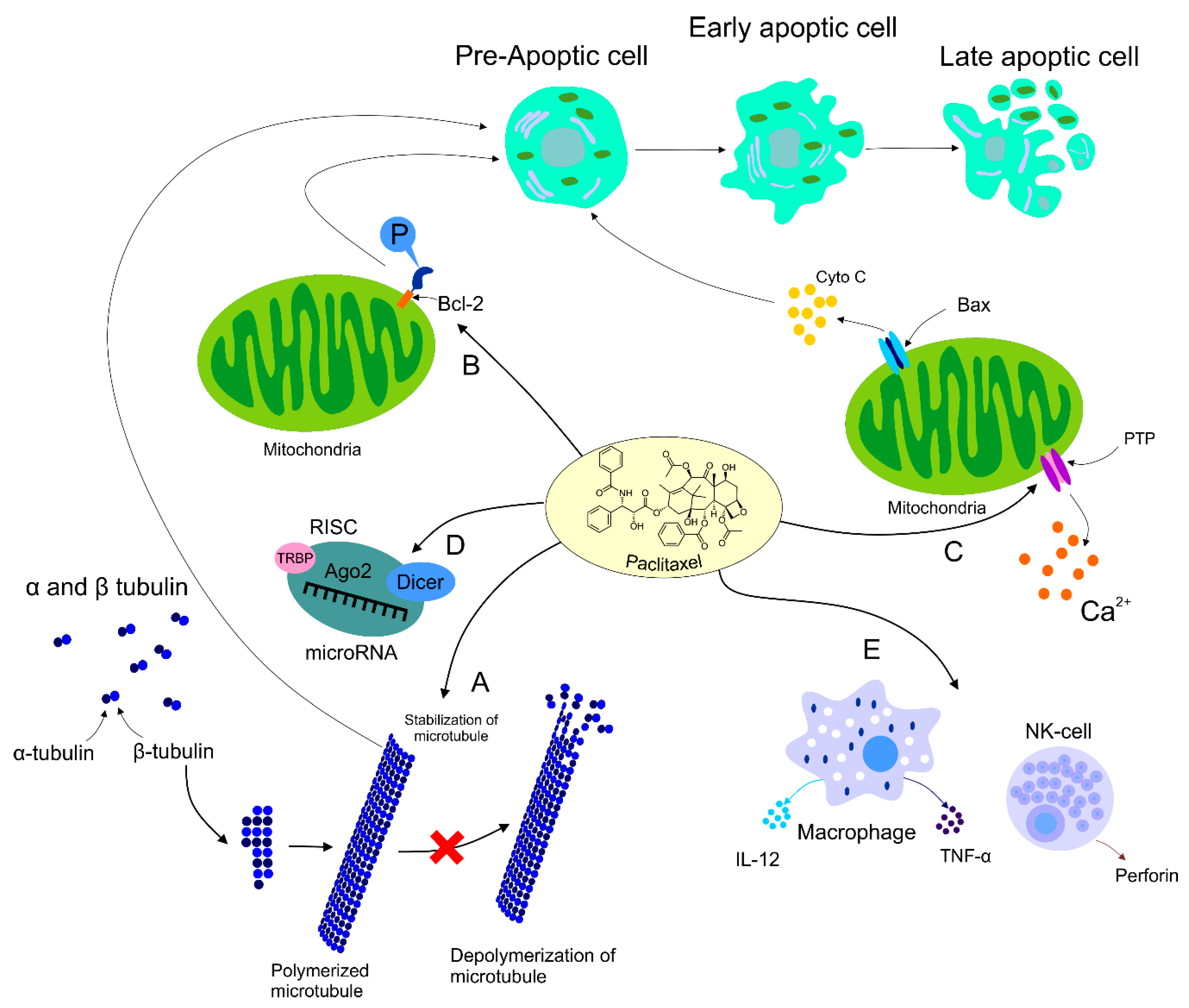
Figure 1. Mechanism of action of PTX. Anti-tumor mechanism of action of PTX leading to stabilization of microtubule, cell arrest, and subsequent apoptosis (A). PTX also causes activation of the immune response contributing to tumor eradication (B). The ability of PTX to inactivate Bcl-2 via phosphorylation of the anti-apoptotic protein resulting in apoptosis (C). Participation of PTX in the regulation of certain miRNAs associated with the modulation of tumor progression (D). Regulation of calcium signaling by PTX results in PTX-induced release of cyto C from the mitochondria and programmed cell death.
2.3. Paclitaxel’s Effect on HER2+ Breast Cancer
The HER2+ subset of BC is more actively biochemically studied as opposed to other subsets. The efficacy of PTX in HER2+ BC patients was inconsistent and contradicting. In 1998, HER2+ BC was found to be biochemically resistant to PTX. The over-expression of HER2 upregulates p21cip1, which inhibits p34cdc2 that is normally activated by PTX in order to induce apoptosis of cancerous cells at the G2/M phase; therefore, the cytotoxic effect of PTX was inhibited [52]. More recent studies conducted on 3121 node-positive postoperative patients showed that the addition of PTX in adjunction to doxorubicin plus cyclophosphamide decreased the rate of recurrence and death significantly upon 10-year follow up [53]. In contrast, out of 46.7% patients responding to taxanes, 65.2% represented HER2+ and 35.5% HER2− tumors [54]. The researcher suggested that PTX works on a signaling transduction pathway specific to HER2+ cancer. PTX activates the tumor suppressant protein p35 and the CDK inhibitor p21WAF1. On the other hand, p21WAF1 is not activated due to the usual activation of p35 since cancer cells lack the presence of this tumor suppressor. Nevertheless, it is suggested that p21WAF1 activation is c-raf-1 dependent since PTX activates c-raf-1, and the accumulation of p21WAF1would not be possible in the case of c-raf-1 depletion [54][55].
2.4. Dose Ranges Administered
A number of studies evaluated different doses of PTX [56]. Table 1 demonstrates the established dose ranges clinically administered to BC patients according to the state and diagnosis of the patient.
Table 1. The administered amount of PTX in correspondence with the patient’s condition and diagnosis.
| Condition | Administration Schedule | Concentration Range | Reference |
|---|---|---|---|
| Adjuvant therapy with doxorubicin (node-positive or high-risk node-negative BC) | Every 3 weeks | 175 mg/m2 IV perfusion over 3 h (4 courses) | [57] |
| Weekly | 80 mg/m2 IV perfusion over 1 h (12 courses) | [58] | |
| Failure of neoadjuvant therapy (MBC or relapse within 6 months of neoadjuvant therapy) | Every 3 weeks | 175 mg/m2 IV perfusion over 3 h | [57] |
| Untreated MBC | Every 3 weeks (max. of 8 cycles) | 200 mg/m2 IV infusion over 3 h + total dose of 480 mg/m2 doxorubicin 25 mg oral prednisone pre-treatment (12 h before treatment) 10 mg intramuscular chlorpheniramine + 300 mg intravenous cimetidine (both 30 min before PTX) |
[59] |
Explanatory notes: +, and/in combination with; h, hours; min, minutes. Abbreviations: BC, breast cancer; MBC, metastatic breast cancer; PTX, Paclitaxel.
Patients pre-treated with anthracyclines did not exhibit a statistically significant response rate compared to those who were not pre-treated with anthracyclines [60]. Using taxanes in conjunction with anthracyclines lowers the risk of disease recurrence and relapse [58]. Although Table 1 shows the clinical dose ranges, a healthcare provider might administer different doses on a different schedule upon clinical assessment. Additionally, Table 2 demonstrates the efficacy of PTX when used in combination with other drugs. As noted, the medical literature does not provide the immunohistochemical or histological profile of the BC, leading to the suggestion that the differential response could be due to the versatile patient body in the trials.
Table 2. Efficacy of PTX as an adjuvant therapy.
| Neoadjuvant Drug Combination | Patient Eligibility | Concentration Range | Efficacy | Reference |
|---|---|---|---|---|
| PTX after Doxorubicin + Cyclophosphamide | Node-positive BC with resected adenocarcinoma | 60 mg/m2 doxorubicin + 600 mg/m2 cyclophosphamide (IV infusion for 30 min to 2 h every 21 days, −4 cycles +4 cycles of 225 mg/m2 PTX (day 1 of each cycle) | PTX + doxorubicin + cyclophosphamide: ↑ DFS by 17% Acceptable toxicity |
[61] |
| PTX + Bevacizumab | MBC patients with/without previous hormonal therapy or adjuvant chemotherapy | 90 mg/m2 PTX (day 1, 8, 15 every 4 weeks) + 10 mg/kg (day 1 and 15) | ↑ progression-free survival (in comparison to PTX alone) ↑ frequency of hypertension, proteinuria, headache, cerebrospinal ischemia |
[62] |
| PTX + Ttrastuzumab | Breast adenocarcinoma patients (tumor no larger than 3 cm, node-negative, min. LVEF of 50%, adequate hematopoietic and liver function) | 80 mg/m2 PTX for 12 weeks + 4 mg/kg trastuzumab (day 1) → 2 mg/kg weekly (12 doses) | 98.7% disease-free survival 99.2% 3-year rate of recurrence-free survival (95%CI) 2.92% of patients reported adverse effects |
[63] |
| PTX + Trastuzumab then post-operative Doxorubicin + Cyclophosphamide | Stage II or III BC patients | Dexamethasone pretreatment (20 mg) + diphenhydramine (12 and 6 h before treatment) and H2-blocker (50 mg) Trastuzumab (one-time loading dose 4 mg/kg) → weekly 2 mg/kg IV infusion for 11 weeks + 175 mg/m2 of IV PTX over 3 h (every 3 weeks, 4 cycles) 2–5 weeks post-op: doxorubicin + cyclophosphamide |
75% clinical response with 18% complete pathologic response Stage 3 tumors responded more than stage 2 tumors |
[64] |
| PTX + rhG-CSF | BC patients (last radiation therapy at least 4 weeks prior to chemotherapy) | 250 mg/m2 of IV PTX (for 24 h every 21 days, dose adjusted to granulocyte and platelet nadirs) 5 μg/kg/d of rhG-CSF (subcutaneously on day 3 through 10/cycle) |
CR—12% of patients PR—50% of patients Inverse correlation between response and median age of patients Minimal toxic effects |
[65] |
Explanatory notes: + plus/and; → followed by; ↑ increase. Abbreviations: BC, breast cancer; CR, complete response; DFS, disease-free survival; LVEF, left ventricular ejection fraction; MBC, metastatic breast cancer; PR, partial response; PTX, Paclitaxel; rhG-CSF, recombinant human granulocyte colony-stimulating factor; h, hours; max., maximum; min., minimum.
2.5. Breast Tumor Resistance to Paclitaxel
Chemoresistance is a major problem of cancer treatment associated with poor response, tumor recurrence, and metastases and represents the leading cause of mortality in BC patients [66][67]. Primary chemoresistance refers to resistance occurring prior to treatment. On the other hand, acquired resistance may develop over time after the chemotherapeutic exposure [67]. Accordingly, treatment with PTX might increase acquired resistance, which leads to chemotherapy failure [68]. Importantly, the mechanisms associated with the challenging and complex nature of chemoresistance [67] are still not clear [69].
Firstly, over-expression of efflux drug proteins is associated with resistance to more than one class of chemotherapeutic agents. The ATP-binding cassette (ABC) superfamily of drug efflux proteins includes P-glycoprotein (P-gp) also known as ABCB1 or MDR-1 [67]. The ABCB gene is involved in the resistance to PTX mediated by over-expression of P-gp, which is consequently associated with the efflux of the drug outside of cells [70]. Importantly, the sensitivity to PTX in PTX-resistant sublines of SK-BR-3 and MCF-7 cells increased significantly but not completely through silencing of ABCB1. Therefore, multiple mechanisms are suggested to be included in PTX resistance in BC cells [69].
Moreover, PTX resistance is associated with spindle assembly checkpoint (SAC) due to the importance of the checkpoint function in PTX sensitivity. Therefore, SAC proteins including Mad2, BubR1 or Aurora A are potentially important markers of PTX resistance [8]. However, suppression of Mad2 and BubR1 in PTX-treated cells eliminated the checkpoint function, which led to the PTX resistance correlating with the reduction of the cyclin-dependent kinase-1 activity [13]. Moreover, over-expression of Aurora kinase A (Aur-A) and FOXM1 was observed in PTX-resistant TNBC cells, suggesting the role of Aur-A in the protection of tumor cells against PTX [71]. Similarly, aberrantly regulated expression of FOXM1 and KIF20A was associated with PTX resistance in MCF-7 cells [72].
Furthermore, alterations in the expression of microtubule-associated proteins (MAPs), such as Tau or MAP4, are also important markers of PTX sensitivity [8]. Microtubule-associated protein Tau may be used as a marker for selection of patients for PTX therapy, as its low expression makes microtubules more vulnerable and BC cells more sensitive to PTX [73].
Moreover, PTX resistance represents an important area of molecular regulation mechanisms connected with changes in miRNA expression that plays a crucial role in chemoresistance to numerous therapy approaches [62][74]. The acquisition of resistance to PTX was identified in an experimental study focusing on the expression of miR-200c-3p. Analyzed miRNA was directly associated with the regulation of SOX2, and over-expression of miR-200c-3p contributed to resistance of BC cells to PTX therapy [14]. Similarly, miR-107 targeted genes related to resistance to Taxol treatment. Upregulation of miR-107 correlated with a decrease in the expression of the oncogene TRIAP1 [75]. Moreover, experimental analysis of MDA-MB-231 clearly proved the inverse correlation between miR-16, which is characterized as a tumor-suppressor element regulating anti-neoplastic events in the cell, and IKBKB. In summary, increased expression of IKBKB corresponded to the chemo-drug resistance to PTX [76]. Furthermore, the participation of over-expressed Lin28 that essentially induced an increase in the expression of Rb and p21 as well as a decrease in the level of let-7 resulted in drug-resistance to Taxol [77]. In addition, recent evidence suggested an interaction between the downregulation of miR-22 and the progression of BC. Using real-time PCR, the lower level of analyzed miR-22 targeting the NRAS oncogene was found to correlate with the reduction of the cancer cells’ sensitivity to PTX [78].
Additionally, increased expression of the actin-binding protein (CapG) promoted PTX resistance in BC cells and xenograft models and was related to the PTX resistance in BC patients through targeting CapG-mediated hyperactivation of the PI3K/Akt pathway [66]. Moreover, increased Y-box protein (YB)-1 levels and decreased EGR1 (early growth response protein 1) mRNA levels correlated with PTX resistance in MDA-MB-231, while a proposed mechanism suggested the biological link between EGR1 and YB-1 and that an increase in YB-1 decreased EGR1 [79]. Interestingly, post-translational modifications such as sumoylation modulated sensitivity to PTX in the BC cell line MCF-7. Data suggested an important role of the post-translational mechanism in the regulation of FOXK2, directly linked to drug resistance in in vitro experiments. Consequently, sumoylation functions as a positive regulator of FOXK2 and its transcriptional activity that subsequently enhances the cytotoxic response to PTX [80].
Taken together, the above-discussed data suggested an important role of various mechanisms associated with ABC, SAC or MAP proteins [67] as well as epigenetic modulation [80] in numerous cellular processes associated with the chemoresistance to PTX. In conclusion, in order to improve the use of chemotherapeutic agents, such as PTX, it is important to clarify the mechanisms underlying the issue of BC chemoresistance [67].
2.6. Paclitaxel’s Side Effects
Importantly, PTX is a chemotherapeutic agent arresting mitosis via microtubule stabilization and consequent induction of apoptosis that is widely used to treat BC. However, the effectiveness of PTX is limited by various side effects associated with its use [81][82].
Major side effects of PTX are hypersensitivity and neuropathies. Due to its poor solubility, PTX has to be formulated in a lipid-based solvent polyoxyl castor oil, also known as Cremophor™, and dehydrated ethanol. However, this vehicle is associated with histamine-mediated hypersensitivity reactions, sensory neuropathy, or impairment of drug delivery as well as limitation of its effectiveness [83][84][85]. PTX hypersensitivity including dyspnea, bronchospasm, urticaria, flushing, erythematous rash, hypotension, angioedema, chest pain, abdominal pain, fever, or rigors is usually visible within the first ten minutes of drug administration. Moreover, PTX hypersensitivity usually results in the immediate halt of the treatment. However, premedication with dexamethasone, diphenhydramine, or cimetidine can reduce hypersensitive reactions. Importantly, reducing the PTX dose by 20% or administration of Amitriptyline may alleviate some of the neuropathic side effects [86]. As mentioned above, peripheral neuropathy is associated with high doses of numerous chemotherapeutic drugs including PTX. In this regard, Vahdat et al. [87] analyzed the reduction of neuropathy after high-dose PTX with oral administration of 10 g glutamine (starting 24 hours after PTX completion), 3 times a day over 4 days. Their clinical data suggested that patients treated with glutamine had significantly decreased levels of severity connected to peripheral neuropathy compared to the placebo control group [87]. Nevertheless, the severity of neuropathy noted in patients depends on the time of the PTX dosage repetition. Patients with weekly administration of PTX showed signs of grade 2, 3, and 4 neuropathy more frequently than patients who were subjected to the drug every three weeks. In addition, patients who received PTX complained of neutropenia, febrile neutropenia, and infection, although the most common side effect was neuropathy [58]. Although the exact mechanism of PTX causing neuropathy is unknown, the administration of Ethosuximide, an anti-epileptic drug and a selective T-type calcium channel blocker, completely reversed the neuropathic symptoms, which suggests that T-type calcium channels are ingrained in the mechanism [88]. The administration of G-CSF (granulocyte colony-stimulating factor) with PTX significantly decreased the severity of neutropenia [86]. Additionally, the establishment of polar micelles of PTX in the plasma compartment causes non-linear pharmacokinetics and drug entrapment. Consequently, the pharmacodynamics of the drug changes, which may cause a risk of systemic toxicities attributed to the substantial increase in its systemic exposure and reduction of its systemic clearance [85].
Moreover, Cremophor™ was also suggested to alter pharamacokinetics of other anticancer drugs, e.g., anthracyclines [89]. Despite premedications with antihistamines and corticosterois to reduce hypersensitivity reactions, this formulation also requires a special infusion set in order to minimize exposure to releasing diphtalates and prolonged infusion time [85].
Additionally, administration of PTX has been associated with cardiotoxicities such as bradyarrythmias, tachyarrythmias, atrioventricular and bundle branch blocks, and cardiac ischemia [90]. However, when combined with doxorubicin, congestive heart failure surfaced [59]. The cardiotoxic effects of PTX are thought to be the result of Cremophor™ hyperstimulation of the histamine receptors in cardiac cells due to the huge influx of histamine in the presence of Cremophor™ [90][91].
2.7. Albumin-Bound Paclitaxel and Comparison with Its Conventional Alternative
Due to the adverse reactions associated with conventional PTX, a need to develop another way it is used is urgently required [83][84][85]. The pharmacokinetics and pharmacodynamics of PTX have been improved by the development of a nanocarrier delivery system. Nab-PTX, which is defined as PTX bound to albumin nanoparticles [92], is a solvent-free preparation allowing PTX to be delivered as a suspension of albumin nanoparticles [93]. Nab-PTX was designed with an aim to improve the therapeutic potential of PTX and to reduce its toxicity and side effects [94]. Importantly, the advantage of nab-PTX is an ability to increase the delivery of albumin to tumors through receptor-mediated transport, also known as transcytosis (shown in Figure 2) [95][96]. Cav-1 is the main component of plasma membrane invaginations (calveolae), and deregulation of tumor Cav-1 is highly implicated in BC. Moreover, loss of stromal Cav-1 plays an important role in disease recurrence and overall worse prognosis of BC patients [97]. Due to the presence of albumin, the binding of nab-PTX to gp60, an albumin receptor on endothelial cells, leads to the activation of caveolin-1 (Cav-1) and formation of calveolae [95]. Consequently, calveolae transport nab-PTX through endothel into the extracellular space including the tumor interstitium [95]. SPARC (secreted protein acidic and rich in cysteine) is defined as an albumin-binding glycoprotein that is frequently over-expressed in cancer cells [98]. Therefore, in the tumor intestitium, SPARC binds to albumin-bound PTX, which facilitates release of PTX near cancer cells and increases the antitumor efficacy of nab-PTX [95]. Nab-PTX has been proven to be 33% more effective than conventional PTX in MX-1 tumor xenografts due to its interaction with SPARC that is over-expressed especially in BC [99]. In addition, nab-PTX does not require premedication, special IV tubing, or a long perfusion time [100].
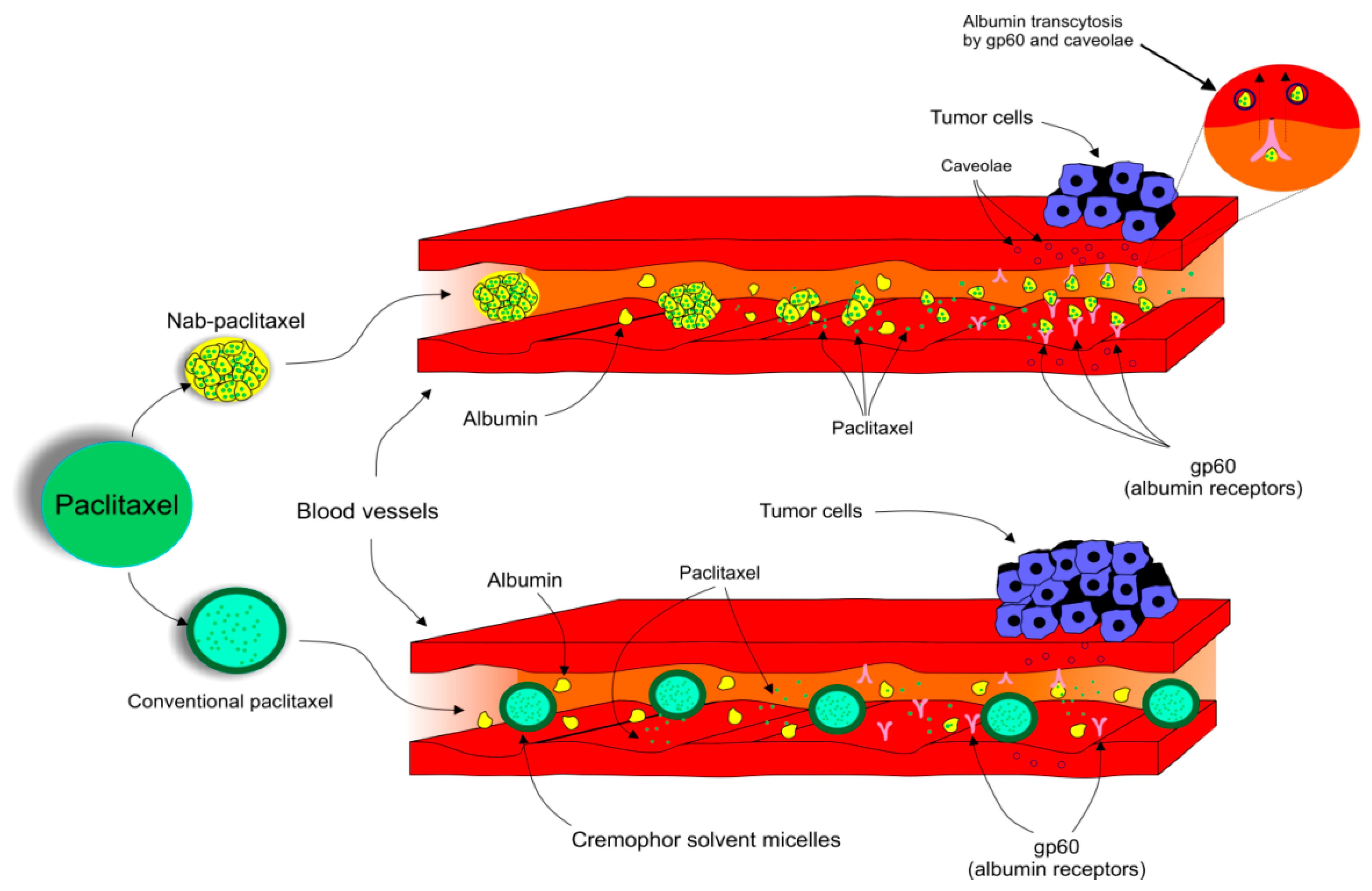
Figure 2. Transcytosis, a receptor-mediated transport of nab-PTX into tumor cells.
A number of clinical trials evaluated the impact of nab-PTX on therapeutic efficacy as well as its association with side effects. Nab-PTX was related to fewer side effects in comparison with conventional PTX, as a lack of hypersensitivity, due to the absence of the Cremophor™ diluent [101]. Weekly neoadjuvant administration of nab-PTX at 100 mg/m2 was associated with good response and tolerability in patients with stage II to IV BC [102]. Additionally, a phase II study of nab-PTX administered weekly in patients with metastatic BC heavily pretreated with taxanes revealed that both 100 mg/m2/weekly and 125 mg/m2/weekly demonstrated the same antitumor activity. However, there were no severe hypersensitivity reactions, and patients with treatment-limiting peripheral neuropathy could be restarted on a reduced dose of nab-PTX after a few weeks delay [100]. Additionally, neoadjuvant therapy with weekly nab-PTX in luminal early BC patients indicated significant drug antitumor activity indicated via a residual cancer burden rate of 0 + 1 with low rates of grade 3-4 toxicity [103]. Moreover, nab-PTX at a dose of 175 mg/m2/3 weeks was associated with less frequent chemotherapy-induced peripheral neuropathy in HER2- metastatic BC patients [104]. A multicenter phase II trial of ABI-007, the first biologically interactive albumin-bound PTX in a nanometer-sized particle, in women with metastatic BC revealed significant anticancer activity, allowing safe administration of high ABI-007 doses without premedication. However, toxicities typical of PTX, including neutropenia (grade 4, 24%), sensory neuropathy (grade 3, 11%), and febrile neutropenia (grade 4, 5%), were observed [105].
Importantly, there is interesting evidence evaluating a comparison of the effectiveness of conventional and nab-PTX. A study using data from an electronic medical record database across the USA revealed that nab-PTX improved clinical effectiveness, demonstrated by a longer time to treatment discontinuation and the time to the next treatment of metastatic BC patients when compared with conventional PTX [16]. Moreover, ABI-007 demonstrated a higher efficacy and safety profile when compared with conventional PTX in women with metastatic BC. Despite that grade 3 neuropathy was more frequent in the ABI-007 group, it was easily manageable (also no hypersensitivity, and grade 4 neutropenia was significantly lower for ABI-007 compared to PTX) [106]. However, Gianni et al. [107] evaluated the ability of nab-PTX to improve the outcomes of early and locally advanced HER2− BC in comparison with PTX in a neoadjuvant setting. Consequently, improvement of pathological complete remission after nab-PTX was not statistically significant [107].
References
- Clatici, V.G.; Voicu, C.; Voaides, C.; Roseanu, A.; Icriverzi, M.; Jurcoane, S. Diseases of civilization—Cancer, diabetes, obesity and acne—The implication of milk, IGF-1 and mTORC1. Mædica 2018, 13, 273–281.
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953.
- Tong, C.W.S.; Wu, M.; Cho, W.C.S.; To, K.K.W. Recent advances in the treatment of breast cancer. Front. Oncol. 2018, 8, 227.
- Abotaleb, M.; Kubatka, P.; Caprnda, M.; Varghese, E.; Zolakova, B.; Zubor, P.; Opatrilova, R.; Kruzliak, P.; Stefanicka, P.; Büsselberg, D. Chemotherapeutic agents for the treatment of metastatic breast cancer: An update. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 101, 458–477.
- Vici, P.; Viola, G.; Rossi, S.; Botti, C.; Vitucci, C.; Sergi, D.; Ferranti, F.R.; Saracca, E.; Di Lauro, L.; Corsetti, S.; et al. Optimal sequence of anthracyclines and taxanes as adjuvant breast cancer treatment. Clin. Ter. 2008, 159, 453–456.
- Kellokumpu-Lehtinen, P.; Tuunanen, T.; Asola, R.; Elomaa, L.; Heikkinen, M.; Kokko, R.; Järvenpää, R.; Lehtinen, I.; Maiche, A.; Kaleva-Kerola, J.; et al. Weekly paclitaxel—An effective treatment for advanced breast cancer. Anticancer Res. 2013, 33, 2623–2627.
- Van Vuuren, R.J.; Visagie, M.H.; Theron, A.E.; Joubert, A.M. Antimitotic drugs in the treatment of cancer. Cancer Chemother. Pharmacol. 2015, 76, 1101–1112.
- McGrogan, B.T.; Gilmartin, B.; Carney, D.N.; McCann, A. Taxanes, microtubules and chemoresistant breast cancer. Biochim. Biophys. Acta 2008, 1785, 96–132.
- Matsuyoshi, S.; Shimada, K.; Nakamura, M.; Ishida, E.; Konishi, N. Bcl-2 phosphorylation has pathological significance in human breast cancer. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 2006, 73, 205–212.
- Pan, Z.; Avila, A.; Gollahon, L. Paclitaxel induces apoptosis in breast cancer cells through different calcium—Regulating mechanisms depending on external calcium conditions. Int. J. Mol. Sci. 2014, 15, 2672–2694.
- Wanderley, C.W.; Colon, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; Leite, C.A.; Pereira, J.A.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1- profile in a TLR4-dependent manner. Cancer Res. 2018, 78, 5891–5900.
- Panis, C.; Pavanelli, W.R. Cytokines as mediators of pain-related process in breast cancer. Mediators Inflamm. 2015, 2015, 129034.
- Sudo, T.; Nitta, M.; Saya, H.; Ueno, N.T. Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer Res. 2004, 64, 2502–2508.
- Chen, J.; Tian, W.; He, H.; Chen, F.; Huang, J.; Wang, X.; Chen, Z. Downregulation of miR-200c-3p contributes to the resistance of breast cancer cells to paclitaxel by targeting SOX2. Oncol. Rep. 2018, 40, 3821–3829.
- Xu, R.; Sato, N.; Yanai, K.; Akiyoshi, T.; Nagai, S.; Wada, J.; Koga, K.; Mibu, R.; Nakamura, M.; Katano, M. Enhancement of Paclitaxel-induced apoptosis by inhibition of mitogen-activated protein Kinase pathway in colon cancer cells. Anticancer Res. 2009, 29, 261–270.
- Mahtani, R.L.; Parisi, M.; Glück, S.; Ni, Q.; Park, S.; Pelletier, C.; Faria, C.; Braiteh, F. Comparative effectiveness of early-line nab-paclitaxel vs. paclitaxel in patients with metastatic breast cancer: A US community-based real-world analysis. Cancer Manag. Res. 2018, 10, 249–256.
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681.
- Walsh, V.; Goodman, J. From taxol to Taxol: The changing identities and ownership of an anti-cancer drug. Med. Anthropol. 2002, 21, 307–336.
- Kumar, P.; Raza, K.; Kaushik, L.; Malik, R.; Arora, S.; Katare, O.P. Role of colloidal drug delivery carriers in Taxane-mediated chemotherapy: A review. Curr. Pharm. Des. 2016, 22, 5127–5143.
- Perdue, R.E.; Hartwell, J.T. Search for plant sources of anticancer drugs. Morris Arbor. Bull 1969, 20, 35–53.
- Walsh, V.; Goodman, J. Cancer chemotherapy, biodiversity, public and private property: The case of the anti-cancer drug taxol. Soc. Sci. Med. 1982 1999, 49, 1215–1225.
- Holton, R.A.; Kim, H.B.; Somoza, C.; Liang, F.; Biediger, R.J.; Boatman, P.D.; Shindo, M.; Smith, C.C.; Kim, S. First total synthesis of taxol. 2. Completion of the C and D rings. J. Am. Chem. Soc. 1994, 116, 1599–1600.
- Stierle, A.; Strobel, G.; Stierle, D. Taxol and taxane production by Taxomyces andreanae, an endophytic fungus of Pacific yew. Science 1993, 260, 214–216.
- Sackett, D.; Fojo, T. Taxanes. Cancer Chemother. Biol. Response Modif. 1997, 17, 59–79.
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667.
- Rowinsky, E.K.; Cazenave, L.A.; Donehower, R.C. Taxol: A novel investigational antimicrotubule agent. JNCI J. Natl. Cancer Inst. 1990, 82, 1247–1259.
- Collins, C.A.; Vallee, R.B. Temperature-dependent reversible assembly of taxol-treated microtubules. J. Cell Biol. 1987, 105, 2847–2854.
- De Brabander, M.; Geuens, G.; Nuydens, R.; Willebrords, R.; De Mey, J. Taxol induces the assembly of free microtubules in living cells and blocks the organizing capacity of the centrosomes and kinetochores. Proc. Natl. Acad. Sci. USA 1981, 78, 5608–5612.
- Jordan, M.A.; Toso, R.J.; Thrower, D.; Wilson, L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc. Natl. Acad. Sci. USA 1993, 90, 9552–9556.
- Jordan, M.A.; Wilson, L. Microtubules and actin filaments: Dynamic targets for cancer chemotherapy. Curr. Opin. Cell Biol. 1998, 10, 123–130.
- Long, B.H.; Fairchild, C.R. Paclitaxel inhibits progression of mitotic cells to G1 phase by interference with spindle formation without affecting other microtubule functions during anaphase and telephase. Cancer Res. 1994, 54, 4355–4361.
- Giannakakou, P.; Robey, R.; Fojo, T.; Blagosklonny, M.V. Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: Molecular determinants of paclitaxel-induced cytotoxicity. Oncogene 2001, 20, 3806–3813.
- Tran, T.-A.; Gillet, L.; Roger, S.; Besson, P.; White, E.; Le Guennec, J.-Y. Non-anti-mitotic concentrations of taxol reduce breast cancer cell invasiveness. Biochem. Biophys. Res. Commun. 2009, 379, 304–308.
- Shetti, D.; Zhang, B.; Fan, C.; Mo, C.; Lee, B.H.; Wei, K. Low dose of paclitaxel combined with XAV939 attenuates metastasis, angiogenesis and growth in breast cancer by suppressing Wnt signaling. Cells 2019, 8, 892.
- Blagosklonny, M.V.; Giannakakou, P.; el-Deiry, W.S.; Kingston, D.G.; Higgs, P.I.; Neckers, L.; Fojo, T. Raf-1/bcl-2 phosphorylation: A step from microtubule damage to cell death. Cancer Res. 1997, 57, 130–135.
- Haldar, S.; Basu, A.; Croce, C.M. Bcl2 is the guardian of microtubule integrity. Cancer Res. 1997, 57, 229–233.
- Ling, Y.H.; Yang, Y.; Tornos, C.; Singh, B.; Perez-Soler, R. Paclitaxel-induced apoptosis is associated with expression and activation of c-Mos gene product in human ovarian carcinoma SKOV3 cells. Cancer Res. 1998, 58, 3633–3640.
- Blagosklonny, M.V.; Fojo, T. Molecular effects of paclitaxel: Myths and reality (a critical review). Int. J. Cancer 1999, 83, 151–156.
- Haldar, S.; Chintapalli, J.; Croce, C.M. Taxol induces bcl-2 phosphorylation and death of prostate cancer cells. Cancer Res. 1996, 56, 1253–1255.
- Ibrado, A.M.; Liu, L.; Bhalla, K. Bcl-xL overexpression inhibits progression of molecular events leading to paclitaxel-induced apoptosis of human acute myeloid leukemia HL-60 cells. Cancer Res. 1997, 57, 1109–1115.
- Blajeski, A.L.; Kottke, T.J.; Kaufmann, S.H. A multistep model for Paclitaxel-induced apoptosis in human breast cancer cell lines. Exp. Cell Res. 2001, 270, 277–288.
- Ofir, R.; Seidman, R.; Rabinski, T.; Krup, M.; Yavelsky, V.; Weinstein, Y.; Wolfson, M. Taxol-induced apoptosis in human SKOV3 ovarian and MCF7 breast carcinoma cells is caspase-3 and caspase-9 independent. Cell Death Differ. 2002, 9, 636–642.
- Varbiro, G.; Veres, B.; Gallyas, F.; Sumegi, B. Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Radic. Biol. Med. 2001, 31, 548–558.
- Kidd, J.F.; Pilkington, M.F.; Schell, M.J.; Fogarty, K.E.; Skepper, J.N.; Taylor, C.W.; Thorn, P. Paclitaxel affects cytosolic calcium signals by opening the mitochondrial permeability transition pore. J. Biol. Chem. 2002, 277, 6504–6510.
- Asghari, F.; Haghnavaz, N.; Shanehbandi, D.; Khaze, V.; Baradaran, B.; Kazemi, T. Differential altered expression of let-7a and miR-205 tumor-suppressor miRNAs in different subtypes of breast cancer under treatment with Taxol. Adv. Clin. Exp. Med. Off. Organ Wroclaw Med. Univ. 2018, 27, 941–945.
- Tao, W.-Y.; Liang, X.-S.; Liu, Y.; Wang, C.-Y.; Pang, D. Decrease of let-7f in low-dose metronomic paclitaxel chemotherapy contributed to upregulation of thrombospondin-1 in breast cancer. Int. J. Biol. Sci. 2015, 11, 48–58.
- Javeed, A.; Ashraf, M.; Riaz, A.; Ghafoor, A.; Afzal, S.; Mukhtar, M.M. Paclitaxel and immune system. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2009, 38, 283–290.
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, M.; Kzhyshkowska, J. Interaction of tumor-associated macrophages and cancer chemotherapy. OncoImmunology 2019, 8, e1596004.
- Emens, L.A.; Jaffee, E.M. Leveraging the activity of tumor vaccines with cytotoxic chemotherapy. Cancer Res. 2005, 65, 8059–8064.
- John, J.; Ismail, M.; Riley, C.; Askham, J.; Morgan, R.; Melcher, A.; Pandha, H. Differential effects of Paclitaxel on dendritic cell function. BMC Immunol. 2010, 11, 14.
- Kubo, M.; Morisaki, T.; Matsumoto, K.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Kuroki, H.; Nakamura, K.; Nakamura, M.; Katano, M. Paclitaxel probably enhances cytotoxicity of natural killer cells against breast carcinoma cells by increasing perforin production. Cancer Immunol. Immunother. CII 2005, 54, 468–476.
- Yu, D.; Jing, T.; Liu, B.; Yao, J.; Tan, M.; McDonnell, T.J.; Hung, M.C. Overexpression of ErbB2 blocks Taxol-induced apoptosis by upregulation of p21Cip1, which inhibits p34Cdc2 kinase. Mol. Cell 1998, 2, 581–591.
- Hayes, D.F.; Thor, A.D.; Dressler, L.G.; Weaver, D.; Edgerton, S.; Cowan, D.; Broadwater, G.; Goldstein, L.J.; Martino, S.; Ingle, J.N.; et al. HER2 and response to paclitaxel in node-positive breast cancer. N. Engl. J. Med. 2007, 357, 1496–1506.
- Baselga, J.; Seidman, A.D.; Rosen, P.P.; Norton, L. HER2 overexpression and paclitaxel sensitivity in breast cancer: Therapeutic implications. Oncol. Williston Park N Y 1997, 11 (Suppl. 2), 43–48.
- Blagosklonny, M.V.; Schulte, T.W.; Nguyen, P.; Mimnaugh, E.G.; Trepel, J.; Neckers, L. Taxol induction of p21WAF1 and p53 requires c-raf-1. Cancer Res. 1995, 55, 4623–4626.
- Winer, E.P.; Berry, D.A.; Woolf, S.; Duggan, D.; Kornblith, A.; Harris, L.N.; Michaelson, R.A.; Kirshner, J.A.; Fleming, G.F.; Perry, M.C.; et al. Failure of higher-dose paclitaxel to improve outcome in patients with metastatic breast cancer: cancer and leukemia group B trial 9342. J. Clin. Oncol. 2004, 22, 2061–2068.
- Spielmann, M. Taxol (Paclitaxel) in patients with metastatic breast carcinoma who have failed prior chemotherapy: Interim results of a multinational study. Oncology 1994, 51, 25–28.
- Sparano, J.A.; Wang, M.; Martino, S.; Jones, V.; Perez, E.A.; Saphner, T.; Wolff, A.C.; Sledge, G.W.; Wood, W.C.; Davidson, N.E. Weekly paclitaxel in the adjuvant treatment of breast cancer. N. Engl. J. Med. 2008, 358, 1663–1671.
- Gianni, L.; Munzone, E.; Capri, G.; Fulfaro, F.; Tarenzi, E.; Villani, F.; Spreafico, C.; Laffranchi, A.; Caraceni, A.; Martini, C. Paclitaxel by 3-hour infusion in combination with bolus doxorubicin in women with untreated metastatic breast cancer: High antitumor efficacy and cardiac effects in a dose-finding and sequence-finding study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1995, 13, 2688–2699.
- Seidman, A.D.; Hudis, C.A.; Albanell, J.; Albanel, J.; Tong, W.; Tepler, I.; Currie, V.; Moynahan, M.E.; Theodoulou, M.; Gollub, M.; et al. Dose-dense therapy with weekly 1-hour paclitaxel infusions in the treatment of metastatic breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1998, 16, 3353–3361.
- Mamounas, E.P.; Bryant, J.; Lembersky, B.; Fehrenbacher, L.; Sedlacek, S.M.; Fisher, B.; Wickerham, D.L.; Yothers, G.; Soran, A.; Wolmark, N. Paclitaxel after doxorubicin plus cyclophosphamide as adjuvant chemotherapy for node-positive breast cancer: Results from NSABP B-28. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 3686–3696.
- Samec, M.; Liskova, A.; Kubatka, P.; Uramova, S.; Zubor, P.; Samuel, S.M.; Zulli, A.; Pec, M.; Bielik, T.; Biringer, K.; et al. The role of dietary phytochemicals in the carcinogenesis via the modulation of miRNA expression. J. Cancer Res. Clin. Oncol. 2019, 145, 1665–1679.
- Tolaney, S.M.; Barry, W.T.; Dang, C.T.; Yardley, D.A.; Moy, B.; Marcom, P.K.; Albain, K.S.; Rugo, H.S.; Ellis, M.; Shapira, I.; et al. Adjuvant Paclitaxel and Trastuzumab for node-negative, HER2-positive breast cancer. N. Engl. J. Med. 2015, 372, 134–141.
- Burstein, H.J.; Harris, L.N.; Gelman, R.; Lester, S.C.; Nunes, R.A.; Kaelin, C.M.; Parker, L.M.; Ellisen, L.W.; Kuter, I.; Gadd, M.A.; et al. Preoperative therapy with trastuzumab and paclitaxel followed by sequential adjuvant doxorubicin/cyclophosphamide for HER2 overexpressing stage II or III breast cancer: A pilot study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 46–53.
- Reichman, B.S.; Seidman, A.D.; Crown, J.P.; Heelan, R.; Hakes, T.B.; Lebwohl, D.E.; Gilewski, T.A.; Surbone, A.; Currie, V.; Hudis, C.A. Paclitaxel and recombinant human granulocyte colony-stimulating factor as initial chemotherapy for metastatic breast cancer. J. Clin. Oncol. 1993, 11, 1943–1951.
- Chi, Y.; Xue, J.; Huang, S.; Xiu, B.; Su, Y.; Wang, W.; Guo, R.; Wang, L.; Li, L.; Shao, Z.; et al. CapG promotes resistance to paclitaxel in breast cancer through transactivation of PIK3R1/P50. Theranostics 2019, 9, 6840–6855.
- Wang, H.; Vo, T.; Hajar, A.; Li, S.; Chen, X.; Parissenti, A.M.; Brindley, D.N.; Wang, Z. Multiple mechanisms underlying acquired resistance to taxanes in selected docetaxel-resistant MCF-7 breast cancer cells. BMC Cancer 2014, 14, 37.
- Wang, Y.; Zhou, Y.; Zheng, Z.; Li, J.; Yan, Y.; Wu, W. Sulforaphane metabolites reduce resistance to paclitaxel via microtubule disruption. Cell Death Dis. 2018, 9, 1134.
- Němcová-Fürstová, V.; Kopperová, D.; Balušíková, K.; Ehrlichová, M.; Brynychová, V.; Václavíková, R.; Daniel, P.; Souček, P.; Kovář, J. Characterization of acquired paclitaxel resistance of breast cancer cells and involvement of ABC transporters. Toxicol. Appl. Pharmacol. 2016, 310, 215–228.
- Childs, S.; Ling, V. The MDR superfamily of genes and its biological implications. Important Adv. Oncol. 1994, 21–36.
- Yang, N.; Wang, C.; Wang, J.; Wang, Z.; Huang, D.; Yan, M.; Kamran, M.; Liu, Q.; Xu, B. Aurora kinase A stabilizes FOXM1 to enhance paclitaxel resistance in triple-negative breast cancer. J. Cell. Mol. Med. 2019, 23, 6442–6453.
- Khongkow, P.; Gomes, A.R.; Gong, C.; Man, E.P.S.; Tsang, J.W.-H.; Zhao, F.; Monteiro, L.J.; Coombes, R.C.; Medema, R.H.; Khoo, U.S.; et al. Paclitaxel targets FOXM1 to regulate KIF20A in mitotic catastrophe and breast cancer paclitaxel resistance. Oncogene 2016, 35, 990–1002.
- Rouzier, R.; Rajan, R.; Wagner, P.; Hess, K.R.; Gold, D.L.; Stec, J.; Ayers, M.; Ross, J.S.; Zhang, P.; Buchholz, T.A.; et al. Microtubule-associated protein tau: A marker of paclitaxel sensitivity in breast cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 8315–8320.
- Magee, P.; Shi, L.; Garofalo, M. Role of microRNAs in chemoresistance. Ann. Transl. Med. 2015, 3, 332.
- Luo, Y.; Hua, T.; You, X.; Lou, J.; Yang, X.; Tang, N. Effects of MiR-107 on the chemo-drug sensitivity of breast cancer cells. Open Med. 2019, 14, 59–65.
- Tang, Q.; Cheng, J.; Cao, X.; Surowy, H.; Burwinkel, B. Blood-based DNA methylation as biomarker for breast cancer: A systematic review. Clin. Epigenetics 2016, 8, 115.
- Lv, K.; Liu, L.; Wang, L.; Yu, J.; Liu, X.; Cheng, Y.; Dong, M.; Teng, R.; Wu, L.; Fu, P.; et al. Lin28 mediates paclitaxel resistance by modulating p21, Rb and Let-7a miRNA in breast cancer cells. PLoS ONE 2012, 7, e40008.
- Song, Y.-K.; Wang, Y.; Wen, Y.-Y.; Zhao, P.; Bian, Z.-J. MicroRNA-22 suppresses breast cancer cell growth and increases Paclitaxel sensitivity by targeting NRAS. Technol. Cancer Res. Treat. 2018, 17.
- Lasham, A.; Mehta, S.Y.; Fitzgerald, S.J.; Woolley, A.G.; Hearn, J.I.; Hurley, D.G.; Ruza, I.; Algie, M.; Shelling, A.N.; Braithwaite, A.W.; et al. A novel EGR-1 dependent mechanism for YB-1 modulation of paclitaxel response in a triple negative breast cancer cell line: EGR-1-dependent mechanism for YB-1 modulation. Int. J. Cancer 2016, 139, 1157–1170.
- Nestal de Moraes, G.; Ji, Z.; Fan, L.Y.-N.; Yao, S.; Zona, S.; Sharrocks, A.D.; Lam, E.W.-F. SUMOylation modulates FOXK2-mediated paclitaxel sensitivity in breast cancer cells. Oncogenesis 2018, 7, 29.
- Zhang, D.; Yang, R.; Wang, S.; Dong, Z. Paclitaxel: new uses for an old drug. Drug Des. Devel. Ther. 2014, 8, 279–284.
- Marupudi, N.I.; Han, J.E.; Li, K.W.; Renard, V.M.; Tyler, B.M.; Brem, H. Paclitaxel: A review of adverse toxicities and novel delivery strategies. Expert Opin. Drug Saf. 2007, 6, 609–621.
- Yamamoto, Y.; Kawano, I. Iwase Nab-paclitaxel for the treatment of breast cancer: Efficacy, safety, and approval. Onco Targets Ther. 2011, 4, 123–136.
- Ma, P.; Mumper, R.J. Paclitaxel nano-delivery systems: A comprehensive review. J. Nanomed. Nanotechnol. 2013, 4, 1000164.
- Vishnu, P.; Roy, V. Safety and efficacy of Nab-Paclitaxel in the treatment of patients with breast cancer. Breast Cancer Basic Clin. Res. 2011, 5, 53–65.
- Walker, F.E. Paclitaxel (TAXOL®): Side effects and patient education issues. Semin. Oncol. Nurs. 1993, 9, 6–10.
- Vahdat, L.; Papadopoulos, K.; Lange, D.; Leuin, S.; Kaufman, E.; Donovan, D.; Frederick, D.; Bagiella, E.; Tiersten, A.; Nichols, G.; et al. Reduction of paclitaxel-induced peripheral neuropathy with glutamine. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001, 7, 1192–1197.
- Flatters, S.J.L.; Bennett, G.J. Ethosuximide reverses paclitaxel- and vincristine-induced painful peripheral neuropathy. Pain 2004, 109, 150–161.
- Ta-Chung, C.; Zyting, C.; Ling-Ming, T.; Tzeon-Jye, C.; Ruey-Kuen, H.; Wei-Shu, W.; Chueh-Chuan, Y.; Muh-Hwa, Y.; Liang-Tsai, H.; Jin-Hwang, L.; et al. Paclitaxel in a novel formulation containing less Cremophor EL as first-line therapy for advanced breast cancer: A phase II trial. Investig. New Drugs 2005, 23, 171–177.
- Rowinsky, E.K.; McGuire, W.P.; Guarnieri, T.; Fisherman, J.S.; Christian, M.C.; Donehower, R.C. Cardiac disturbances during the administration of taxol. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1991, 9, 1704–1712.
- Bristow, M.R.; Sageman, W.S.; Scott, R.H.; Billingham, M.E.; Bowden, R.E.; Kernoff, R.S.; Snidow, G.H.; Daniels, J.R. Acute and chronic cardiovascular effects of doxorubicin in the dog: The cardiovascular pharmacology of drug-induced histamine release. J. Cardiovasc. Pharmacol. 1980, 2, 487–515.
- Hoffman, R.M.; Bouvet, M. Nanoparticle albumin-bound-paclitaxel: A limited improvement under the current therapeutic paradigm of pancreatic cancer. Expert Opin. Pharmacother. 2015, 16, 943–947.
- Roy, V.; LaPlant, B.R.; Gross, G.G.; Bane, C.L.; Palmieri, F.M.; North Central Cancer Treatment Group. Phase II trial of weekly nab (nanoparticle albumin-bound)-paclitaxel (nab-paclitaxel) (Abraxane) in combination with gemcitabine in patients with metastatic breast cancer (N0531). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2009, 20, 449–453.
- Desai, N.; Trieu, V.; Yao, Z.; Louie, L.; Ci, S.; Yang, A.; Tao, C.; De, T.; Beals, B.; Dykes, D.; et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 1317–1324.
- Iglesias, J. Nab-Paclitaxel (Abraxane®): An albumin-bound cytotoxic exploiting natural delivery mechanisms into tumors. Breast Cancer Res. BCR 2009, 11, S21.
- Aapro, M.S.; Von Minckwitz, G. Molecular basis for the development of novel taxanes in the treatment of metastatic breast cancer. Eur. J. Cancer Suppl. 2008, 6, 3–11.
- Zhao, Y.; Lv, F.; Chen, S.; Wang, Z.; Zhang, J.; Zhang, S.; Cao, J.; Wang, L.; Cao, E.; Wang, B.; et al. Caveolin-1 expression predicts efficacy of weekly nab-paclitaxel plus gemcitabine for metastatic breast cancer in the phase II clinical trial. BMC Cancer 2018, 18, 1019.
- Zhu, A.; Yuan, P.; Du, F.; Hong, R.; Ding, X.; Shi, X.; Fan, Y.; Wang, J.; Luo, Y.; Ma, F.; et al. SPARC overexpression in primary tumors correlates with disease recurrence and overall survival in patients with triple negative breast cancer. Oncotarget 2016, 7, 76628–76634.
- Desai, N.; Trieu, V.; Damascelli, B.; Soon-Shiong, P. SPARC expression correlates with tumor response to albumin-bound Paclitaxel in head and neck cancer patients. Transl. Oncol. 2009, 2, 59–64.
- Blum, J.L.; Savin, M.A.; Edelman, G.; Pippen, J.E.; Robert, N.J.; Geister, B.V.; Kirby, R.L.; Clawson, A.; O’Shaughnessy, J.A. Phase II study of weekly albumin-bound Paclitaxel for patients with metastatic breast cancer heavily pretreated with Taxanes. Clin. Breast Cancer 2007, 7, 850–856.
- Montana, M.; Ducros, C.; Verhaeghe, P.; Terme, T.; Vanelle, P.; Rathelot, P. Albumin-bound Paclitaxel: The benefit of this new formulation in the treatment of various cancers. J. Chemother. 2011, 23, 59–66.
- Matsui, A.; Tatibana, A.; Suzuki, N.; Hirata, M.; Oishi, Y.; Hamaguchi, Y.; Murata, Y.; Nagayama, A.; Iwata, Y.; Okamoto, Y. Evaluation of efficacy and safety of upfront weekly nanoparticle albumin-bound Paclitaxel for HER2-negative breast cancer. Anticancer Res. 2017, 37, 6481–6488.
- Martín, M.; Chacón, J.I.; Antón, A.; Plazaola, A.; García-Martínez, E.; Seguí, M.A.; Sánchez-Rovira, P.; Palacios, J.; Calvo, L.; Esteban, C.; et al. Neoadjuvant therapy with weekly nanoparticle albumin-bound Paclitaxel for luminal early breast cancer patients: Results from the NABRAX study (GEICAM/2011-02), a multicenter, non-randomized, phase II trial, with a companion biomarker analysis. Oncologist 2017, 22, 1301–1308.
- Takashima, T.; Kawajiri, H.; Nishimori, T.; Tei, S.; Nishimura, S.; Yamagata, S.; Tokunaga, S.; Mizuyama, Y.; Sunami, T.; Tezuka, K.; et al. Safety and efficacy of low-dose nanoparticle albumin-bound Paclitaxel for HER2-negative metastatic breast cancer. Anticancer Res. 2018, 38, 379–383.
- Ibrahim, N.K.; Samuels, B.; Page, R.; Doval, D.; Patel, K.M.; Rao, S.C.; Nair, M.K.; Bhar, P.; Desai, N.; Hortobagyi, G.N. Multicenter phase II trial of ABI-007, an albumin-bound paclitaxel, in women with metastatic breast cancer. J. Clin. Oncol. 2005, 23, 6019–6026.
- Gradishar, W.J.; Tjulandin, S.; Davidson, N.; Shaw, H.; Desai, N.; Bhar, P.; Hawkins, M.; O’Shaughnessy, J. Phase III trial of nanoparticle albumin-bound Paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J. Clin. Oncol. 2005, 23, 7794–7803.
- Gianni, L.; Mansutti, M.; Anton, A.; Calvo, L.; Bisagni, G.; Bermejo, B.; Semiglazov, V.; Thill, M.; Chacon, J.I.; Chan, A.; et al. Comparing neoadjuvant nab-Paclitaxel vs. Paclitaxel both followed by Anthracycline regimens in women with ERBB2/HER2-negative breast cancer-the Evaluating Treatment With Neoadjuvant Abraxane (ETNA) trial: A randomized phase 3 clinical trial. JAMA Oncol. 2018, 4, 302–308.
This entry is offline, you can click here to edit this entry!