3.2. Genetic Markers
The advent of new genome analysis technologies played a major role in the process of understanding tumor pathogenesis and heterogeneity. In a large study conducted among 489 CCA patients from 10 countries, Jusakul et al. [
133] analyzed genetic features of CCA, including the whole genome sequencing (WGS) (
n = 71), and DNA methylation (
n = 138) assessment. In the WGS analysis, a total of 1,309,932 mutations were detected across 71 tumor samples, including 4541 nonsilient single nucleotide variations (SNVs) and 1251 nonsilient indels. On average, each tumor had 82 nonsilient mutations, including 64 SNV and 18 indels. Fluke-positive CCAs were significantly more plentiful in somatic mutations comparing with Fluke-negative tumors (median of 4700 vs. 3143 per tumor, respectively). Fluke infection was also correlated with poorer survival. Based on the results of their analysis, the authors suggest dividing CAA cases into 4 molecular clusters:
Cluster 1 was characterized by mostly Fluke-positive CCAs with hypermethylation of promoter CpG islands, enrichment of TP53, ARID1A, and BRCA1/2 DNA repair associated (BCRA1/2) mutations with reduced expression of Tet methylcytosine dioxygenase 1 (TET1), and enhanced expression of enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) and Erb-B2 receptor tyrosine kinase 2 (ERBB2) amplification.
Cluster 2 was also enriched in TP53 mutations and ERBB2 amplification, CTNNB1, Wnt family member 5B (WNT5B), and AKT serine/threonine kinase 1 (AKT1). It consisted of both Fluke-positive and Fluke-negative CCAs and represented a low level of methylation.
Cluster 3 showed specific upregulation of immune system genes, including immune checkpoint genes (programmed death receptor 1 (PD-1), programmed cell death 1 ligand 2 (PD-L2), and B- and T-lymphocyte-associated protein (BTLA)) and pathways related to the costimulation of T lymphocytes. Similar to Cluster 2, it was also characterized by a low methylation level.
Cluster 4 was characterized by enrichment of BRCA1 associated protein 1 (BAP1) and isocitrate dehydrogenase (NADP(+)) 1/isocitrate dehydrogenase (NADP(+)) 2 (IDH1/2) mutations and fibroblast growth factor receptor (FGFR) aberrations with upregulated expression of FGFR1, FGFR2, FGFR3 and FGFR4. Similar to Cluster 1 its level of methylation was high, but, inversely, the methylation phenotype included CpG shore hypermethylation instead of CpG island hypermethylation, suggesting distinct mutational pathways.
Additionally, Clusters 1 and 2 were mostly represented by extrahepatic tumors, while Clusters 3 and 4 were characterized by intrahepatic malignancies almost entirely. Clusters 3 and 4 were associated with significantly better overall survival.
In another study, Lowery et al. [
134] investigated molecular profiling of intrahepatic and extrahepatic CCA. An analysis carried out among CCA samples of 195 patients showed that in intrahepatic CCA the most commonly seen aberrations were
: IDH1 (30%),
ARID1A (23%),
BAP1 (20%),
TP53 (20%), and
FGFR2 gene fusions (14%). In the case of extrahepatic CCA, the most commonly found aberrations were: KRAS proto-oncogene, GTPase (
KRAS), SMAD family member 4 (
SMAD4), and serine/threonine kinase 11 (
STK11) alterations. In addition,
CDKN2A/B and
ERBB2 gene alterations were correlated with reduced overall survival and time to progression on first-line chemotherapy. Forty-seven percent of the patients showed somatic alterations with potential therapeutic value and thus 16% of the patients were enrolled in clinical trials molecular therapies.
Li et al. [
135] carried out a study aimed at exploring the biological functions and prognostic biomarkers involved in CCA through transcriptional analysis. Thirty-three samples were obtained from CCA patients and 8 normal tissue samples. They discovered a total of 1463 differentially expressed genes, of which 267 were significantly upregulated and the remaining 1196 were significantly downregulated. According to Gene Ontology (GO) analysis, upregulated genes were enriched in ‘cadherin binding in cell-cell adhesion’, ‘extracellular matrix organization’ and ‘cell-cell adherens junctions’, while the downregulated ones were enriched in ‘oxidation-reduction process’, ‘extracellular exosomes’ and ‘blood microparticles’. Twenty-one of the genes were defied as hub genes including 8 upregulated genes and 13 downregulated genes. For all of the 21 hub genes, AUC was >0.900. Among upregulated hub genes, the expression level of
CDK1, marker of proliferation Ki-67 (
MKI67), DNA topoisomerase II alpha (
TOP2A), and protein regulator of cytokinesis 1
(PRC1) were significantly negatively correlated to overall survival, while no similar correlation was found in remaining hub genes. Additionally, among downregulated hub genes, the expression of acyl-CoA oxidase 1 (
ACOX1), apolipoprotein A2 (
APOA2), apolipoprotein B (
APOB), fibrinogen alpha chain (
FGA), and fibrinogen gamma chain
(FGG) were negatively correlated with the tumor stage of CCA patients. The authors suggest that
CDK1,
MKI67,
TOP2A, and
PRC1 could be used as prognostic biomarkers of CCA. The summary of detection of the biomarkers is showed in
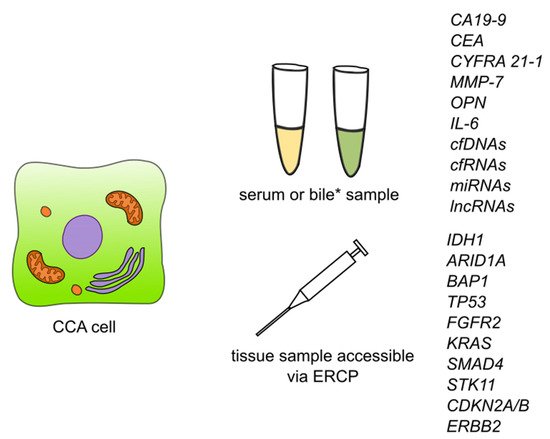
Figure 3.
Figure 3. Summary of detection of biomarkers and some of the genetic markers for cholangiocarcinoma. Abbreviations: CA19-9—carbohydrate antigen 19-9/cancer antigen 19-9; CEA—carcinoembryonic antigen; CYFRA 21-1—cytokeratin fragment antigen 21-1; MMP-7—metalloproteinase 7; IL-6—interleukin 6; cfDNAs—circulating free DNAs; cfRNAs—circulating free RNAs; lncRNAs—long non-coding RNAs; ERCP—endoscopic retrograde cholangiopancreatography; IDH1—isocitrate dehydrogenase (NADP(+)) 1; ARID1A—AT-rich interaction domain 1A; BAP1—BRCA1 associated protein 1; TP53—tumor protein P53; FGFR2—fibroblast growth factor receptor 2; KRAS—KRAS proto-oncogene, GTPase; SMAD4—SMAD family member 4; STK11—serine/threonine kinase 11; CDKN2A/B—cyclin dependent kinase inhibitor 2A/cyclin dependent kinase inhibitor 2B; ERBB2—Erb-B2 receptor tyrosine kinase 2. * Detection of CCA biomarkers in bile has so far been described for CEA and cfDNAs.