Primary cilia are non-motile plasma membrane extrusions that display a variety of receptors and mechanosensors. Loss of function results in ciliopathies, which have been strongly linked with congenital heart disease, as well as abnormal development and function of most organ systems.
- primary cilia
- congenital heart disease
- ciliopathy
- cardiomyopathy
- heart failure
1. Primary Cilia
1.1. Cilia Structure and Components
Primary cilia are extrusions of the plasma membrane that display a variety of receptors and mechanosensors. The core structure is an axoneme of nine doublet microtubules that extend from a basal body, and they are therefore referred to as “9 + 0” cilia. This distinguishes them from motile “9 + 2” cilia, which have an additional two dynein-associated central microtubules, permitting motion.[1]
As primary cilia do not intrinsically have associated ribosomes, they instead rely on the intraflagellar transport (IFT) system to ferry receptors and other proteins into and out of the cilium.[2] This system is capable of bidirectional movement along the length of the flagella, between the outer doublet of microtubules and the flagellar membrane.[3][4] IFT proteins, especially Ift88, are often knockout targets in cilia research, as their inactivation results in the absence of primary cilia in the affected cell.[5][6]
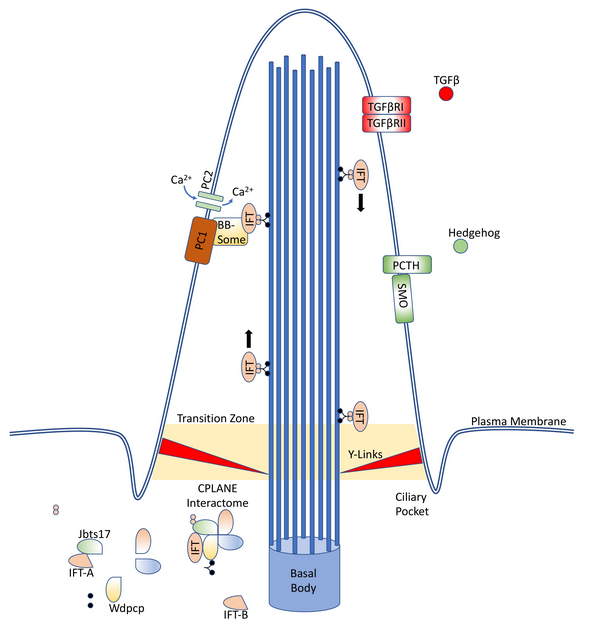
1.2. Ciliopathies
For classification purposes, first-order ciliopathies are those diseases which occur due to a mutation in genes required for the proper assembly, maintenance, or function of the cilia or the related centriole; second-order ciliopathies occur due to dysregulation of further upstream factors, such as the nuclear transcription factors Atf3, Tsc22d4, and Cbx5.[13][14] There are at least 300–1000 first-order, and many more second-order, genes.[13][15][16]
1.3. Primary Cilia Locations
2. Primary Cilia in Acquired Heart Disease
2.1. Acquired Valvular Heart Disease
The importance of proper cilia function in the embryonic heart has been well established.[34][6][35][36] In a comprehensive analysis of over 87,000 mutagenized mouse fetuses, Li et al. identified 61 genes in which mutations were capable of producing echocardiographically identifiable congenital heart defects, and 35 of these genes encoded either motile or primary cilia proteins. An additional 16 genes were involved in cilia-transduced cell signaling, and 10 regulated vesicular trafficking, which is necessary for proper cilia function.[34]
2.2. Fibrosis
In addition to myxomatous degeneration of the valve, patients with MVP also show progressive left ventricular fibrosis. Cardiac fibrosis is an excessive production and deposition of scar tissue, often a result of conditions such as hypertension or diabetes mellitus, and can lead to increased tissue stiffness, cardiomyocyte atrophy, and arrhythmias.[40][41] The fibrosis observed with MVP is more significant than that seen in patients with primary mitral valve regurgitation from a non-MVP etiology, which may suggest a common cause for both excessive fibrosis and MVP.[42]
In addition to native cardiac fibroblast proliferation, endothelial-mesenchymal transition (EndMT) is now recognized as an important source of fibroblasts for perivascular and subendocardial fibrosis.[44] Knockdown of Ift88 in endothelial cells, which results in the absence of primary cilia on these cells, appears to be insufficient to directly induce EndMT in vivo but may prime these cells for EndMT in response to lower stress than would otherwise be required.[45][46]
2.3. Vascular Pathology and Cilia
2.4. Ventricular Remodeling and Recovery
One possible mechanism appears to be via ciliary extracellular-like vesicles (cELVs).[61] These vesicles are released from cilia under normal circumstances and at increased rates under fluid shear stress. Blocking ciliary proteins necessary for cELV production using short hairpin RNA (shRNA) prevents cELV production and results in left ventricular hypertrophy, decreasing left ventricular ejection fraction, and, eventually, low blood pressure and cardiovascular collapse.[61][62]
2.5. Congenital Heart Disease and Late-Onset Heart Failure
This entry is adapted from the peer-reviewed paper 10.3390/cells11060960
References
- Peter Satir; Lotte B. Pedersen; Søren T. Christensen; The primary cilium at a glance. Journal of Cell Science 2010, 123, 499-503, 10.1242/jcs.050377.
- Andrew M Fry; Michelle J Leaper; Richard Bayliss; The primary cilium. Organogenesis 2013, 10, 62-68, 10.4161/org.28910.
- K G Kozminski; K A Johnson; P Forscher; J L Rosenbaum; A motility in the eukaryotic flagellum unrelated to flagellar beating.. Proceedings of the National Academy of Sciences 1993, 90, 5519-5523, 10.1073/pnas.90.12.5519.
- Michinori Toriyama; University of Washington Center for Mendelian Genomics; Chanjae Lee; S Paige Taylor; Ivan Duran; Daniel H Cohn; Ange-Line Bruel; Jacqueline M Tabler; Kevin Drew; Marcus Kelly; et al. The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nature Genetics 2016, 48, 648-656, 10.1038/ng.3558.
- Vincent Z. Luu; Albert Z. Luu; Biswajit Chowdhury; Omar Elbardisy; Yi Pan; Mohammed Al-Omran; Adrian Quan; Hwee Teoh; David A. Hess; Subodh Verma; et al. Disruption of endothelial cell intraflagellar transport protein 88 exacerbates doxorubicin-induced cardiotoxicity. Life Sciences 2020, 260, 118216, 10.1016/j.lfs.2020.118216.
- Marc August Willaredt; Karin Gorgas; Humphrey A R Gardner; Kerry L Tucker; Multiple essential roles for primary cilia in heart development. Cilia 2012, 1, 23-23, 10.1186/2046-2530-1-23.
- Kelsey Moore; Reece Moore; Christina Wang; Russell A. Norris; Tugging at the Heart Strings: The Septin Cytoskeleton in Heart Development and Disease. Journal of Cardiovascular Development and Disease 2020, 7, 3, 10.3390/jcdd7010003.
- Dominique Douguet; Amanda Patel; Eric Honoré; Structure and function of polycystins: insights into polycystic kidney disease. Nature Reviews Nephrology 2019, 15, 412-422, 10.1038/s41581-019-0143-6.
- K Kojima; I Sakai; A Hasegawa; H Niiya; T Azuma; Y Matsuo; N Fujii; M Tanimoto; S Fujita; FLJ10849, a septin family gene, fuses MLL in a novel leukemia cell line CNLBC1 derived from chronic neutrophilic leukemia in transformation with t(4;11)(q21;q23). Leukemia 2004, 18, 998-1005, 10.1038/sj.leu.2403334.
- Dimitrios Angelis; Elias T. Spiliotis; Septin Mutations in Human Cancers. Frontiers in Cell and Developmental Biology 2016, 4, 122, 10.3389/fcell.2016.00122.
- Ayae Kinoshita; Makoto Kinoshita; Haruhiko Akiyama; Hidekazu Tomimoto; Ichiro Akiguchi; Sharad Kumar; Makoto Noda; Jun Kimura; Identification of Septins in Neurofibrillary Tangles in Alzheimer's Disease. The American Journal of Pathology 1998, 153, 1551-1560, 10.1016/s0002-9440(10)65743-4.
- Italo A. Cavini; Diego A. Leonardo; Higor V. D. Rosa; Danielle K. S. V. Castro; Humberto D’Muniz Pereira; Napoleão F. Valadares; Ana P. U. Araujo; Richard C. Garratt; The Structural Biology of Septins and Their Filaments: An Update. Frontiers in Cell and Developmental Biology 2021, 9, 1, 10.3389/fcell.2021.765085.
- Marta Lovera; Jens Lüders; The ciliary impact of nonciliary gene mutations. Trends in Cell Biology 2021, 31, 876-887, 10.1016/j.tcb.2021.06.001.
- Gabrielle Wheway; UK10K Consortium; Miriam Schmidts; Dorus A. Mans; Katarzyna Szymanska; Thanh-Minh T. Nguyen; Hilary Racher; Ian G. Phelps; Grischa Toedt; Julie Kennedy; et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nature Cell Biology 2015, 17, 1074-1087, 10.1038/ncb3201 [doi].
- John van Dam; SYSCILIA Study Group; Gabrielle Wheway; Gisela G Slaats; Martijn Huynen; Rachel H Giles; The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia 2013, 2, 1, 10.1186/2046-2530-2-7 [doi].
- Teunis J. P. van Dam; Julie Kennedy; Robin van der Lee; Erik de Vrieze; Kirsten A. Wunderlich; Suzanne Rix; Gerard W. Dougherty; Nils J. Lambacher; Chunmei Li; Victor L. Jensen; et al. CiliaCarta: An integrated and validated compendium of ciliary genes. PLoS ONE 2019, 14, e0216705, 10.1371/journal.pone.0216705.
- Clarisse Delvallée; Samuel Nicaise; Manuela Antin; Anne‐Sophie Leuvrey; Elsa Nourisson; Carmen C. Leitch; Georgios Kellaris; Corinne Stoetzel; Véronique Geoffroy; Sophie Scheidecker; et al. A BBS1 SVA F retrotransposon insertion is a frequent cause of Bardet‐Biedl syndrome. Clinical Genetics 2020, 99, 318-324, 10.1111/cge.13878.
- Jarema J. Malicki; Colin A. Johnson; The Cilium: Cellular Antenna and Central Processing Unit. Trends in Cell Biology 2016, 27, 126-140, 10.1016/j.tcb.2016.08.002.
- Ganesh V. Pusapati; Jennifer Kong; Bhaven B. Patel; ArunKumar Krishnan; Andreas Sagner; Maia Kinnebrew; James Briscoe; L. Aravind; Rajat Rohatgi; CRISPR Screens Uncover Genes that Regulate Target Cell Sensitivity to the Morphogen Sonic Hedgehog. Developmental Cell 2017, 44, 113-129.e8, 10.1016/j.devcel.2017.12.003.
- David K. Breslow; Sascha Hoogendoorn; Adam R. Kopp; David W. Morgens; Brandon K. Vu; Margaret C. Kennedy; Kyuho Han; Amy Li; Gaelen T. Hess; Michael C. Bassik; et al. A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies. Nature Genetics 2018, 50, 460-471, 10.1038/s41588-018-0054-7.
- Lasse Jonsgaard Larsen; Lisbeth Birk Møller; Crosstalk of Hedgehog and mTORC1 Pathways. Cells 2020, 9, 2316, 10.3390/cells9102316.
- Fiona Bangs; Kathryn V. Anderson; Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harbor Perspectives in Biology 2016, 9, a028175, 10.1101/cshperspect.a028175.
- Manuela Morleo; Brunella Franco; The Autophagy-Cilia Axis: An Intricate Relationship.. Cells 2019, 8, 905, 10.3390/cells8080905.
- Sarah C. Goetz; Kathryn Anderson; The primary cilium: a signalling centre during vertebrate development. Nature Reviews Genetics 2010, 11, 331-344, 10.1038/nrg2774.
- Ji-Eun Bae; Gil Myung Kang; Se Hee Min; Doo Sin Jo; Yong-Keun Jung; Keetae Kim; Min-Seon Kim; Dong-Hyung Cho; Primary cilia mediate mitochondrial stress responses to promote dopamine neuron survival in a Parkinson’s disease model. Cell Death & Disease 2019, 10, 1-15, 10.1038/s41419-019-2184-y.
- Junguee Lee; Ki Cheol Park; Hae Joung Sul; Hyun Jung Hong; Kun-Ho Kim; Jukka Kero; Minho Shong; Loss of primary cilia promotes mitochondria-dependent apoptosis in thyroid cancer. Scientific Reports 2021, 11, 1-15, 10.1038/s41598-021-83418-3.
- Kimberly F. Atkinson; Rinzhin T. Sherpa; Surya M. Nauli; The Role of the Primary Cilium in Sensing Extracellular pH.. Cells 2019, 8, 704, 10.3390/cells8070704.
- Collins I; Wann A.K.T; Isabella Collins; A.K.T Wann; Regulation of the Extracellular Matrix by Ciliary Machinery. Cells 2020, 9, 278, 10.3390/cells9020278.
- Elisa Villalobos; Alfredo Criollo; Gabriele Schiattarella; Francisco Altamirano; Kristin M. French; Herman May; Nan Jiang; Ngoc Uyen Nhi Nguyen; Diego Romero; Juan Carlos Roa; et al. Fibroblast Primary Cilia Are Required for Cardiac Fibrosis. Circulation 2019, 139, 2342-2357, 10.1161/circulationaha.117.028752.
- Xinhua Li; Shuting Yang; Vishwa Deepak; Zahra Chinipardaz; Shuying Yang; Identification of Cilia in Different Mouse Tissues. Cells 2021, 10, 1623, 10.3390/cells10071623.
- Hua Zhang; Dan Chalothorn; James E Faber; Collateral Vessels Have Unique Endothelial and Smooth Muscle Cell Phenotypes. International Journal of Molecular Sciences 2019, 20, 3608, 10.3390/ijms20153608.
- Katelynn A. Toomer; Mengyao Yu; Diana Fulmer; Lilong Guo; Kelsey S. Moore; Reece Moore; Ka’La D. Drayton; Janiece Glover; Neal Peterson; Sandra Ramos-Ortiz; et al. Primary cilia defects causing mitral valve prolapse. Science Translational Medicine 2019, 11, 1, 10.1126/scitranslmed.aax0290.
- Sarbjot Kaur; Sue R. McGlashan; Marie-Louise Ward; Evidence of primary cilia in the developing rat heart. Cilia 2018, 7, 1-7, 10.1186/s13630-018-0058-z.
- You Li; Nikolai T. Klena; George C. Gabriel; Xiaoqin Liu; Andrew J. Kim; Kristi Lemke; Yu Chen; Bishwanath Chatterjee; William Devine; Rama Rao Damerla; et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 2015, 521, 520-524, 10.1038/nature14269.
- Kylia Williams; Jason Carson; Cecilia Lo; Genetics of Congenital Heart Disease. Biomolecules 2019, 9, 879, 10.3390/biom9120879.
- Diana Fulmer; Katelynn Toomer; Lilong Guo; Kelsey Moore; Janiece Glover; Reece Moore; Rebecca Stairley; Glenn Lobo; Xiaofeng Zuo; Yujing Dang; et al. Defects in the Exocyst-Cilia Machinery Cause Bicuspid Aortic Valve Disease and Aortic Stenosis. Circulation 2019, 140, 1331-1341, 10.1161/circulationaha.119.038376.
- Francesca N. Delling; Ramachandran S. Vasan; Epidemiology and Pathophysiology of Mitral Valve Prolapse. Circulation 2014, 129, 2158-2170, 10.1161/circulationaha.113.006702.
- Ronen Durst; Kimberly Sauls; David S. Peal; Annemarieke DeVlaming; Katelynn Toomer; Maire Leyne; Monica Salani; Michael Talkowski; Harrison Brand; Maëlle Perrocheau; et al. Mutations in DCHS1 cause mitral valve prolapse. Nature 2015, 525, 109-113, 10.1038/nature14670.
- Katelynn A Toomer; Diana Fulmer; Lilong Guo; Alex Drohan; Neal Peterson; Paige Swanson; Brittany Brooks; Rupak Mukherjee; Simon Body; Joshua H. Lipschutz; et al. A role for primary cilia in aortic valve development and disease. Developmental Dynamics 2017, 246, 625-634, 10.1002/dvdy.24524.
- Andrew Leask; Getting to the Heart of the Matter. Circulation Research 2015, 116, 1269-1276, 10.1161/circresaha.116.305381.
- Robert A. Levine; Michael Jerosch-Herold; Roger J. Hajjar; Mitral Valve Prolapse. Journal of the American College of Cardiology 2018, 72, 835-837, 10.1016/j.jacc.2018.07.006.
- Danai Kitkungvan; Faisal Nabi; Raymond J. Kim; Robert O. Bonow; Ahmad Khan; Jiaqiong Xu; Stephen H. Little; Miguel A. Quinones; Gerald M. Lawrie; William A. Zoghbi; et al. Myocardial Fibrosis in Patients With Primary Mitral Regurgitation With and Without Prolapse. Journal of the American College of Cardiology 2018, 72, 823-834, 10.1016/j.jacc.2018.06.048.
- Marcin Dobaczewski; Judith J. de Haan; Nikolaos G. Frangogiannis; The Extracellular Matrix Modulates Fibroblast Phenotype and Function in the Infarcted Myocardium. Journal of Cardiovascular Translational Research 2012, 5, 837-847, 10.1007/s12265-012-9406-3.
- Elisabeth M Zeisberg; Oleg Tarnavski; Michael Zeisberg; Adam L Dorfman; Julie R McMullen; Erika Gustafsson; Anil Chandraker; Xueli Yuan; William T Pu; Anita B Roberts; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nature Medicine 2007, 13, 952-961, 10.1038/nm1613.
- Shweta Singh; Mohamed Adam; Pratiek N. Matkar; Antoinette Bugyei-Twum; Jean-Francois Desjardins; Hao H. Chen; Hien Nguyen; Hannah Bazinet; David Michels; Zongyi Liu; et al. Endothelial-specific Loss of IFT88 Promotes Endothelial-to-Mesenchymal Transition and Exacerbates Bleomycin-induced Pulmonary Fibrosis. Scientific Reports 2020, 10, 1-14, 10.1038/s41598-020-61292-9.
- Anastasia D. Egorova; Padmini P.S.J. Khedoe; Marie-José T.H. Goumans; Bradley K. Yoder; Surya M. Nauli; Peter Ten Dijke; Robert E. Poelmann; Beerend P. Hierck; Lack of Primary Cilia Primes Shear-Induced Endothelial-to-Mesenchymal Transition. Circulation Research 2011, 108, 1093-1101, 10.1161/CIRCRESAHA.110.231860.
- Milos Spasic; Christopher R. Jacobs; Primary cilia: Cell and molecular mechanosensors directing whole tissue function. Seminars in Cell & Developmental Biology 2017, 71, 42-52, 10.1016/j.semcdb.2017.08.036.
- Vincent Z. Luu; Biswajit Chowdhury; Mohammed Al-Omran; David A. Hess; Subodh Verma; Role of endothelial primary cilia as fluid mechanosensors on vascular health. Atherosclerosis 2018, 275, 196-204, 10.1016/j.atherosclerosis.2018.06.818.
- Surya M. Nauli; Yoshifumi Kawanabe; John J. Kaminski; William J. Pearce; Donald E. Ingber; Jing Zhou; Endothelial Cilia Are Fluid Shear Sensors That Regulate Calcium Signaling and Nitric Oxide Production Through Polycystin-1. Circulation 2008, 117, 1161-1171, 10.1161/circulationaha.107.710111.
- Rajasekharreddy Pala; Maha Jamal; Qamar Alshammari; Surya M. Nauli; The Roles of Primary Cilia in Cardiovascular Diseases.. Cells 2018, 7, 233, 10.3390/cells7120233.
- Caroline Cheng; Dennie Tempel; Rien Van Haperen; Arjen Van Der Baan; Frank Grosveld; Mat Daemen; Rob Krams; Rini De Crom; Atherosclerotic Lesion Size and Vulnerability Are Determined by Patterns of Fluid Shear Stress. Circulation 2006, 113, 2744-2753, 10.1161/circulationaha.105.590018.
- Kim van der Heiden; Beerend Hierck; Rob Krams; Rini de Crom; Caroline Cheng; Martin Baiker; Mathieu J.B.M. Pourquie; Fanneke E. Alkemade; Marco C. DeRuiter; Adriana C. Gittenberger-De Groot; et al. Endothelial primary cilia in areas of disturbed flow are at the base of atherosclerosis. Atherosclerosis 2008, 196, 542-550, 10.1016/j.atherosclerosis.2007.05.030.
- Lina C. Espinha; David Hoey; Paulo Rui Fernandes; Helder Rodrigues; Christopher R. Jacobs; Oscillatory fluid flow influences primary cilia and microtubule mechanics. Cytoskeleton 2014, 71, 435-445, 10.1002/cm.21183.
- Colin Dinsmore; Jeremy F Reiter; Endothelial primary cilia inhibit atherosclerosis. EMBO reports 2016, 17, 156-166, 10.15252/embr.201541019.
- Christine Patch; Judith Charlton; Paul J. Roderick; Martin C. Gulliford; Use of Antihypertensive Medications and Mortality of Patients With Autosomal Dominant Polycystic Kidney Disease: A Population-Based Study. American Journal of Kidney Diseases 2011, 57, 856-862, 10.1053/j.ajkd.2011.01.023.
- Shakila Abdul-Majeed; Surya M. Nauli; Dopamine Receptor Type 5 in the Primary Cilia Has Dual Chemo- and Mechano-Sensory Roles. Hypertension 2011, 58, 325-331, 10.1161/hypertensionaha.111.172080.
- Chunyu Zeng; Pedro A. Jose; Dopamine Receptors. Hypertension 2010, 57, 11-17, 10.1161/hypertensionaha.110.157727.
- N. Frey; E.N. Olson; Cardiac Hypertrophy: The Good, the Bad, and the Ugly. Annual Review of Physiology 2003, 65, 45-79, 10.1146/annurev.physiol.65.092101.142243 092101.142243.
- Norifumi Takeda; Ichiro Manabe; Yuichi Uchino; Kosei Eguchi; Sahohime Matsumoto; Satoshi Nishimura; Takayuki Shindo; Motoaki Sano; Kinya Otsu; Paige Snider; et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. Journal of Clinical Investigation 2010, 120, 254-265, 10.1172/jci40295.
- Mary J. Roman; Antonello Ganau; Pier Sergio Saba; Riccardo Pini; Thomas G. Pickering; Richard B. Devereux; Impact of Arterial Stiffening on Left Ventricular Structure. Hypertension 2000, 36, 489-494, 10.1161/01.hyp.36.4.489.
- Ann-Kathrin Volz; Alina Frei; Viola Kretschmer; António M. De Jesus Domingues; Rene F. Ketting; Marius Ueffing; Karsten Boldt; Eva-Maria Krämer-Albers; Helen L. May-Simera; Bardet-Biedl syndrome proteins modulate the release of bioactive extracellular vesicles. Nature Communications 2021, 12, 1-16, 10.1038/s41467-021-25929-1.
- Ashraf M. Mohieldin; Rajasekharreddy Pala; Rinzhin T. Sherpa; Madhawi Alanazi; Ashwaq Alanazi; Kiumars Shamloo; Amir Ahsan; Wissam A. AbouAlaiwi; James J. Moresco; John R. Yates; et al. Proteomic Identification Reveals the Role of Ciliary Extracellular‐Like Vesicle in Cardiovascular Function. Advanced Science 2020, 7, 1903140, 10.1002/advs.201903140.
- Thomas P Graham; Yvonne D Bernard; Beverly G Mellen; David Celermajer; Helmut Baumgartner; Frank Cetta; Heidi M Connolly; William R Davidson; Mikael Dellborg; Elyse Foster; et al. Long-term outcome in congenitally corrected transposition of the great arteries: A multi-institutional study. Journal of the American College of Cardiology 2000, 36, 255-261, 10.1016/s0735-1097(00)00682-3.
- Robert B. Hinton; Stephanie M. Ware; Heart Failure in Pediatric Patients With Congenital Heart Disease. Circulation Research 2017, 120, 978-994, 10.1161/circresaha.116.308996.
- Kambiz Norozi; Armin Wessel; Valentin Alpers; Jan Ole Arnhold; Siegfried Geyer; Monika Zoege; Reiner Buchhorn; Incidence and Risk Distribution of Heart Failure in Adolescents and Adults With Congenital Heart Disease After Cardiac Surgery. The American Journal of Cardiology 2006, 97, 1238-1243, 10.1016/j.amjcard.2005.10.065.
- Myrthe E. Menting; Judith A.A.E. Cuypers; Petra Opić; Elisabeth M.W.J. Utens; Maarten Witsenburg; Annemien E. Van Den Bosch; Ron T. van Domburg; Folkert J. Meijboom; Eric Boersma; Ad J.J.C. Bogers; et al. The Unnatural History of the Ventricular Septal Defect. Journal of the American College of Cardiology 2015, 65, 1941-1951, 10.1016/j.jacc.2015.02.055.