: We have witnessed noteworthy progress in our understanding of prostate cancer over
the past decades. This basic knowledge has been translated into efficient diagnostic and treatment
approaches leading to the improvement in patient survival. However, the molecular pathogenesis of
prostate cancer appears to be complex, and histological findings often do not provide an accurate
assessment of disease aggressiveness and future course. Moreover, we also witness tremendous racial
disparity in prostate cancer incidence and clinical outcomes necessitating a deeper understanding
of molecular and mechanistic bases of prostate cancer. Biological research heavily relies on model
systems that can be easily manipulated and tested under a controlled experimental environment.
Over the years, several cancer cell lines have been developed representing diverse molecular subtypes
of prostate cancer. In addition, several animal models have been developed to demonstrate the
etiological molecular basis of the prostate cancer. In recent years, patient-derived xenograft and 3-D
culture models have also been created and utilized in preclinical research. This review is an attempt
to succinctly discuss existing information on the cellular and molecular progression of prostate cancer.
We also discuss available model systems and their tested and potential utility in basic and preclinical
prostate cancer research.
- prostate cancer
- research model
- oncogenes
- tumor suppressor genes
1. Definition
Prostate cancer (PCa) is the most commonly diagnosed malignancy and the second leading cause of cancer-related death in men in the United States. It is estimated that PCa will afflict approximately 191,930 men and cause nearly 33,330 deaths this year in the United States alone [1]. Notably, PCa incidence and associated mortality are nearly two-thirds and over two times higher, respectively, in African-American (AA) men compared to their Caucasian-American (CA) counterparts [2,3]. PCa follows a defined pattern of cellular progression but exhibits diverse molecular pathobiology making it one of most highly heterogeneous cancers [4,5]. The prostate-specific antigen (PSA) test is the primary detection tool for PCa screening. However, due to the lack of accuracy and specificity, the usefulness of PSA for PCa diagnosis has been questioned [6,7,8]. Most PCa patients are generally subjected to localized radical prostatectomy, radiation therapy, proton beam therapy, and cryosurgery after the initial diagnosis [9,10,11]. However, for patients with metastatic disease or recurrent cancer with locoregional and distant metastases, androgen-deprivation therapy (ADT) or castration therapy is considered the primary line of treatment [12]. Unfortunately, despite the initial outstanding therapeutic response, most PCa patients treated with ADT eventually have the relapse of PCa in a highly aggressive and therapy-resistant form leading to poor clinical outcomes [13,14].
2. Cellular and Molecular Progression of Prostate Cancer
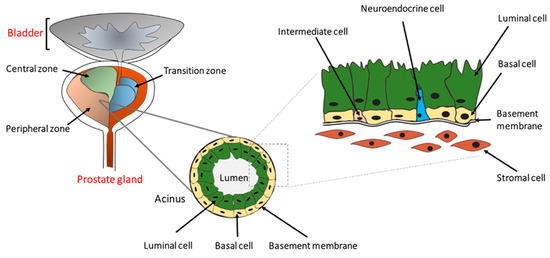
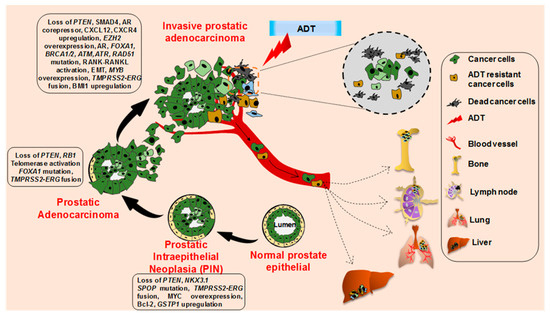
3. Prostate Cancer Research Models
| Cell Line | Origin | Doubling Time | AR | PSA | Markers | Cyto-Keratin | Source | Refs. |
|---|---|---|---|---|---|---|---|---|
| Non-cancerous prostate epithelial cell lines | ||||||||
| RWPE-1 | NPEC in peripheral zone | 120 h | + | + | p53, Rb | 8, 18 | ATCC | [88,89] |
| BPH-1 | Primary prostatic tissue | 35 h | − | − | p53, BAX, PTEN, p21 | 8, 18, 19 | ACCEGEN, Creative Bioarray, DSMZ | [90] |
| pRNS-1-1 | radical prostatectomy | 72 h | − | − | PTEN | 5, 8 | NCI and Stanford University | [91] |
| RC77N/E | Non-malignant tissue of a PCa patient | No report | + | − | NKX3.1, p16 | 8 | Tuskegee University | [92] |
| HprEpC | Normal human prostate | No report | + | + | Cytokeratin 18 | 14, 18, 19 | Cell applications, iXcells Biotechnologies, EZ biosystem | [93] |
| Hormone sensitive | ||||||||
| LNCaP | lymph node metastatic | 28–60 h | + | + | WT p53, PTEN loss, vimentin, PAP, CBP, negative desmin | 8, 18, 20 | ATCC, Creative Bioarray, ACCEGEN, SIGMA | [94] |
| LAPC-4 | lymph node metastatic from an androgen insensitive patient | 72 h | + | + | p53 mutation | 5, 8, 18 | ATCC * | [95] |
| LAPC-9 | bone metastasis from a patient with ADT | No report | + | + | Ki67, PTEN loss | 5 | ATCC * | [96] |
| VCaP | metastatic tumor | 51 h | + | + | p53 mutation, Rb, PAP, PTEN | 8, 18 | ATCC, SIGMA, ACCEGEN | [97] |
| MDA-PCa 2a/2b | bone metastasis from an African-American male | 82–93 h/42–73 h | + | + | WT p53, p21, Rb, Bcl-2 | 5, 8, 18 | ATCC | [98] |
| LuCaP 23.1 | lymph node and liver metastatic | 11–21 days | + | + | 5α-reductase type I, WT PTEN | No report | University of Washington | [99] |
| RC-77T/E | Radical prostatectomy from an African-American patient | No report | + | + | p16, NKX3.1, β-catenin, α-actinin-1, filamin-A | 8 | Tuskegee University | [92] |
| Castration resistant | ||||||||
| PC-3 | lumbar vertebral metastasis | 33 h | − | − | PTEN loss, no p53 expression, TGF-α, EGFR, transferrin receptor | 7, 8, 18, 19 | ATCC, SIGMA, ACCEGEN, Creative Bioarray | [100] |
| DU-145 | Brain metastasis | 34 h | − | − | TGF-α/β, EGFR, IGF-1, EGF | 5, 7, 8, 18 | ATCC, ACCEGEN | [101] |
| C4-2/C4-2B | mouse vertebral metastasis LNCaP cell xenograft | 48 h | + | + | p53, PTEN loss, marker chromosome m1 | 8 | ATCC | [102,103] |
| 22Rv1 | CWR22R xenograft derivative | 35–40 h | + | + | kallikrien-like serine protease, AR splice variant | 8, 18 | ATCC, SIGMA, ACCEGEN, Creative Bioarray | [104] |
| ARCaP | ascites fluid of a patient with advanced metastatic disease | No report | + | + | EGFR, c-erb B2/neu, c-erb B3, bombesin, serotonin | 8, 18 | Novicure Biotechnology | [105] |
3.1. Cell Line Models
3.1.1. Non-Cancerous Prostate Epithelial Cell Lines
RWPE-1
BPH-1
pRNS-1-1
RC-77N/E
HprEpC
3.1.2. Prostate Cancer Cell Lines
Castration-Sensitive
LNCaP
LAPC-4
LAPC-9
RWPE-2
VCaP
MDA-PCa 2a/2b
LuCaP 23.1
RC-77T/E
12T-7f
Castration-Resistant Cell Lines
Androgen-Receptor Expressing
C4-2/C4-2B
22Rv1
Androgen-Receptor Non-Expressing
PC-3
DU-145
ARCaP
3.2. Genetically Engineered Mouse Models of Prostate Cancer
3.2.1. TRAMP
3.2.2. LADY
3.2.3. Pten Deficient Mice
3.2.4. Ptenpc−/−Smad4pc−/−
3.2.5. Hi/Lo-Myc
3.2.6. MPAKT
3.3. Patient Tumor-Derived Models
3.3.1. Three-Dimensional (3-D) Organoid Cultures
3.3.2. Patient-Derived Xenografts (PDX)
| Model | Advantages | Limitations | Sources |
|---|---|---|---|
| 3D-organoid |
|
|
Primary prostate cancer patient-derived tissue |
| PDX |
|
|
Primary prostate cancer patient-derived tissue, CrownBio, The Jackson Laboratory |
3.4. Other Models
3.4.1. Rat Models
3.4.2. Zebrafish Model
4. Conclusions and Future Outlook
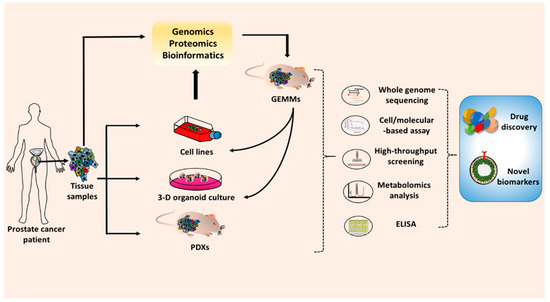
This entry is adapted from the peer-reviewed paper 10.3390/cancers12092651