Epigenetics encompasses a group of dynamic, reversible, and heritable modifications that occur within cells that are independent of gene mutations. These alterations are highly influenced by the environment, from the environment that surrounds the human being to the internal microenvironments located within tissues and cells. The ways that pigenetic modifications promote the initiation of the tumorigenic process have been widely demonstrated. Similarly, it is well known that carcinogenesis is supported and prompted by a strong proinflammatory environment. At the same time, cancer cells can alter their epigenetic profile to generate a positive loop in the promotion of the inflammatory process. Therefore, an in-depth understanding of the epigenetic networks between the tumor microenvironment and cancer cells might highlight new targetable mechanisms that could prevent tumor progression.
- epigenetics
- carcinogenesis
- inflammation
- tumor microenvironment
1. Introduction: The Complexity of Carcinogenesis
2. Overview of Epigenetic Modifications
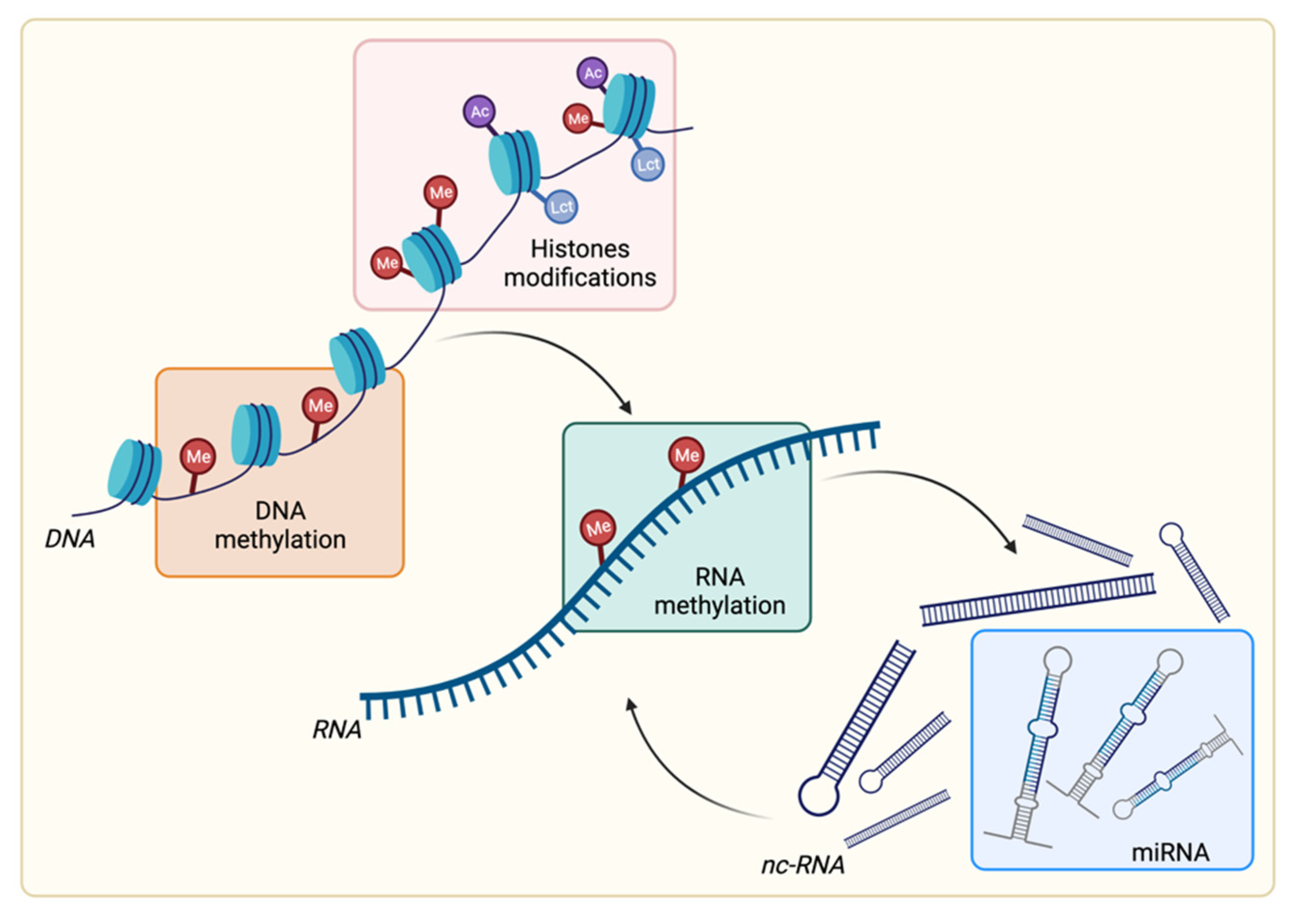
2.1. DNA Methylation
2.2. RNA Methylation
2.3. Histone Posttranslational Modifications
2.4. Noncoding RNA: Focus on microRNA
This entry is adapted from the peer-reviewed paper 10.3390/cancers14051221
References
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558.
- Easwaran, H.; Tsai, H.-C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727.
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; da Silva Santos, R.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176.
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90.
- Chen, Q.W.; Zhu, X.Y.; Li, Y.Y.; Meng, Z.Q. Epigenetic regulation and cancer (review). Oncol. Rep. 2014, 31, 523–532.
- Lu, Y.; Chan, Y.-T.; Tan, H.-Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79.
- Cedar, H.; Bergman, Y. Programming of DNA methylation patterns. Annu. Rev. Biochem. 2012, 81, 97–117.
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322.
- Ziller, M.J.; Müller, F.; Liao, J.; Zhang, Y.; Gu, H.; Bock, C.; Boyle, P.; Epstein, C.B.; Bernstein, B.E.; Lengauer, T.; et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011, 7, e1002389.
- Ramsahoye, B.H.; Biniszkiewicz, D.; Lyko, F.; Clark, V.; Bird, A.P.; Jaenisch, R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242.
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492.
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590.
- Panneerdoss, S.; Eedunuri, V.K.; Yadav, P.; Timilsina, S.; Rajamanickam, S.; Viswanadhapalli, S.; Abdelfattah, N.; Onyeagucha, B.C.; Cui, X.; Lai, Z.; et al. Cross-talk among writers, readers, and erasers of m6A regulates cancer growth and progression. Sci. Adv. 2018, 4, eaar8263.
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA Methylation in Cancer and Aging. Cancer Res. 2016, 76, 3446–3450.
- Welner, S.A.; Koty, Z.C.; Boksa, P. Chromaffin cell grafts to rat cerebral cortex reverse lesion-induced memory deficits. Brain Res. 1990, 527, 163–166.
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236.
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874.
- Chan, H.L.; Morey, L. Emerging Roles for Polycomb-Group Proteins in Stem Cells and Cancer. Trends Biochem. Sci. 2019, 44, 688–700.
- Issa, J.-P. Aging and epigenetic drift: A vicious cycle. J. Clin. Investig. 2014, 124, 24–29.
- Ma, Y.; Chen, Y.; Petersen, I. Expression and promoter DNA methylation of MLH1 in colorectal cancer and lung cancer. Pathol. Res. Pract. 2017, 213, 333–338.
- Botezatu, A.; Iancu, I.V.; Plesa, A.; Manda, D.; Popa, O.; Bostan, M.; Mihaila, M.; Albulescu, A.; Fudulu, A.; Vladoiu, S.V.; et al. Methylation of tumour suppressor genes associated with thyroid cancer. Cancer Biomark. 2019, 25, 53–65.
- Yokoyama, T.; Takehara, K.; Sugimoto, N.; Kaneko, K.; Fujimoto, E.; Okazawa-Sakai, M.; Okame, S.; Shiroyama, Y.; Yokoyama, T.; Teramoto, N.; et al. Lynch syndrome-associated endometrial carcinoma with MLH1 germline mutation and MLH1 promoter hypermethylation: A case report and literature review. BMC Cancer 2018, 18, 576.
- Bonora, M.; Missiroli, S.; Perrone, M.; Fiorica, F.; Pinton, P.; Giorgi, C. Mitochondrial Control of Genomic Instability in Cancer. Cancers 2021, 13, 1914.
- Hughes, L.A.E.; Melotte, V.; de Schrijver, J.; de Maat, M.; Smit, V.T.H.B.M.; Bovée, J.V.M.G.; French, P.J.; van den Brandt, P.A.; Schouten, L.J.; de Meyer, T.; et al. The CpG island methylator phenotype: What’s in a name? Cancer Res. 2013, 73, 5858–5868.
- Ma, S.; Chen, C.; Ji, X.; Liu, J.; Zhou, Q.; Wang, G.; Yuan, W.; Kan, Q.; Sun, Z. The interplay between m6A RNA methylation and noncoding RNA in cancer. J. Hematol. Oncol. 2019, 12, 121.
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95.
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.-M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.-L.; Song, S.-H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29.
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624.
- Sun, T.; Wu, R.; Ming, L. The role of m6A RNA methylation in cancer. Biomed. Pharmacother. 2019, 112, 108613.
- Sadakierska-Chudy, A.; Filip, M. A comprehensive view of the epigenetic landscape. Part II: Histone post-translational modification, nucleosome level, and chromatin regulation by ncRNAs. Neurotox. Res. 2015, 27, 172–197.
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080.
- Voss, A.K.; Thomas, T. Histone Lysine and Genomic Targets of Histone Acetyltransferases in Mammals. Bioessays 2018, 40, e1800078.
- Gayther, S.A.; Batley, S.J.; Linger, L.; Bannister, A.; Thorpe, K.; Chin, S.F.; Daigo, Y.; Russell, P.; Wilson, A.; Sowter, H.M.; et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000, 24, 300–303.
- Fei, H.-J.; Zu, L.-D.; Wu, J.; Jiang, X.-S.; Wang, J.-L.; Chin, Y.E.; Fu, G.-H. PCAF acts as a gastric cancer suppressor through a novel PCAF-p16-CDK4 axis. Am. J. Cancer Res. 2016, 6, 2772–2786.
- Wan, J.; Xu, W.; Zhan, J.; Ma, J.; Li, X.; Xie, Y.; Wang, J.; Zhu, W.-G.; Luo, J.; Zhang, H. PCAF-mediated acetylation of transcriptional factor HOXB9 suppresses lung adenocarcinoma progression by targeting oncogenic protein JMJD6. Nucleic Acids Res. 2016, 44, 10662–10675.
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400.
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628.
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809.
- Sarkar, R.; Banerjee, S.; Amin, S.A.; Adhikari, N.; Jha, T. Histone deacetylase 3 (HDAC3) inhibitors as anticancer agents: A review. Eur. J. Med. Chem. 2020, 192, 112171.
- Zee, B.M.; Levin, R.S.; Xu, B.; LeRoy, G.; Wingreen, N.S.; Garcia, B.A. In vivo residue-specific histone methylation dynamics. J. Biol. Chem. 2010, 285, 3341–3350.
- Bai, L.; Charvin, G.; Siggia, E.D.; Cross, F.R. Nucleosome-depleted regions in cell-cycle-regulated promoters ensure reliable gene expression in every cell cycle. Dev. Cell 2010, 18, 544–555.
- Collings, C.K.; Anderson, J.N. Links between DNA methylation and nucleosome occupancy in the human genome. Epigenet. Chromatin 2017, 10, 18.
- Soufi, A.; Donahue, G.; Zaret, K.S. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 2012, 151, 994–1004.
- Onder, T.T.; Kara, N.; Cherry, A.; Sinha, A.U.; Zhu, N.; Bernt, K.M.; Cahan, P.; Marcarci, B.O.; Unternaehrer, J.; Gupta, P.B.; et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature 2012, 483, 598–602.
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810.
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134.
- Béguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013, 23, 677–692.
- Hodges, C.; Kirkland, J.G.; Crabtree, G.R. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026930.
- Berns, K.; Sonnenblick, A.; Gennissen, A.; Brohée, S.; Hijmans, E.M.; Evers, B.; Fumagalli, D.; Desmedt, C.; Loibl, S.; Denkert, C.; et al. Loss of ARID1A Activates ANXA1, which Serves as a Predictive Biomarker for Trastuzumab Resistance. Clin. Cancer Res. 2016, 22, 5238–5248.
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184.e7.
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580.
- Latham, T.; Mackay, L.; Sproul, D.; Karim, M.; Culley, J.; Harrison, D.J.; Hayward, L.; Langridge-Smith, P.; Gilbert, N.; Ramsahoye, B.H. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 2012, 40, 4794–4803.
- Genders, A.J.; Martin, S.D.; McGee, S.L.; Bishop, D.J. A physiological drop in pH decreases mitochondrial respiration, and HDAC and Akt signaling, in L6 myocytes. Am. J. Physiol. Cell Physiol. 2019, 316, C404–C414.
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108.
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529.
- Lovat, F.; Fassan, M.; Sacchi, D.; Ranganathan, P.; Palamarchuk, A.; Bill, M.; Karunasiri, M.; Gasparini, P.; Nigita, G.; Distefano, R.; et al. Knockout of both miR-15/16 loci induces acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2018, 115, 13069–13074.
- Young, L.E.; Moore, A.E.; Sokol, L.; Meisner-Kober, N.; Dixon, D.A. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol. Cancer Res. 2012, 10, 167–180.
- Bonci, D.; Coppola, V.; Musumeci, M.; Addario, A.; Giuffrida, R.; Memeo, L.; D’Urso, L.; Pagliuca, A.; Biffoni, M.; Labbaye, C.; et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat. Med. 2008, 14, 1271–1277.
- Sun, C.-Y.; She, X.-M.; Qin, Y.; Chu, Z.-B.; Chen, L.; Ai, L.-S.; Zhang, L.; Hu, Y. miR-15a and miR-16 affect the angiogenesis of multiple myeloma by targeting VEGF. Carcinogenesis 2013, 34, 426–435.
- Huang, E.; Liu, R.; Chu, Y. miRNA-15a/16: As tumor suppressors and more. Future Oncol. 2015, 11, 2351–2363.
- Hünten, S.; Siemens, H.; Kaller, M.; Hermeking, H. The p53/microRNA network in cancer: Experimental and bioinformatics approaches. Adv. Exp. Med. Biol. 2013, 774, 77–101.
- Ji, Q.; Hao, X.; Meng, Y.; Zhang, M.; Desano, J.; Fan, D.; Xu, L. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer 2008, 8, 266.
- Warne, M.S.; Connell, D.W.; Hawker, D.W. Comparison of the critical concentration and critical volume hypotheses to model non-specific toxicity of individual compounds. Toxicology 1991, 66, 187–195.
- Xia, H.; Ng, S.S.; Jiang, S.; Cheung, W.K.C.; Sze, J.; Bian, X.-W.; Kung, H.-F.; Lin, M.C. miR-200a-mediated downregulation of ZEB2 and CTNNB1 differentially inhibits nasopharyngeal carcinoma cell growth, migration and invasion. Biochem. Biophys. Res. Commun. 2010, 391, 535–541.
- Pecot, C.V.; Rupaimoole, R.; Yang, D.; Akbani, R.; Ivan, C.; Lu, C.; Wu, S.; Han, H.-D.; Shah, M.Y.; Rodriguez-Aguayo, C.; et al. Tumour angiogenesis regulation by the miR-200 family. Nat. Commun. 2013, 4, 2427.
- Bui, T.V.; Mendell, J.T. Myc: Maestro of MicroRNAs. Genes Cancer 2010, 1, 568–575.
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287–314.
- Kent, O.A.; Chivukula, R.R.; Mullendore, M.; Wentzel, E.A.; Feldmann, G.; Lee, K.H.; Liu, S.; Leach, S.D.; Maitra, A.; Mendell, J.T. Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway. Genes Dev. 2010, 24, 2754–2759.
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589.
- Lujambio, A.; Calin, G.A.; Villanueva, A.; Ropero, S.; Sánchez-Céspedes, M.; Blanco, D.; Montuenga, L.M.; Rossi, S.; Nicoloso, M.S.; Faller, W.J.; et al. A microRNA DNA methylation signature for human cancer metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 13556–13561.
- Lujambio, A.; Ropero, S.; Ballestar, E.; Fraga, M.F.; Cerrato, C.; Setién, F.; Casado, S.; Suarez-Gauthier, A.; Sanchez-Cespedes, M.; Git, A.; et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007, 67, 1424–1429.
- Neves, R.; Scheel, C.; Weinhold, S.; Honisch, E.; Iwaniuk, K.M.; Trompeter, H.-I.; Niederacher, D.; Wernet, P.; Santourlidis, S.; Uhrberg, M. Role of DNA methylation in miR-200c/141 cluster silencing in invasive breast cancer cells. BMC Res. Notes 2010, 3, 219.