Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Allergy
With respect to structural and functional cardiac disorders, heart failure (HF) is divided into HF with reduced ejection fraction (HFrEF) and HF with preserved ejection fraction (HFpEF). Oxidative stress contributes to the development of both HFrEF and HFpEF. Identification of a broad spectrum of reactive oxygen species (ROS)-induced pathways in preclinical models has provided new insights about the importance of ROS in HFrEF and HFpEF development.
- reactive oxygen species
- protein kinases
- heart failure
- NO
- adenosine monophosphate activated protein kinase
- mitogen-activated protein kinase
- superoxide dismutase
- xanthine oxidase
- nitric oxide synthase
- guanylyl cyclase
1. Introduction
Being a by-product of aerobic metabolism, ROS are abundant in the cells of the myocardium and, if the balance between ROS production and antioxidant systems is impaired, they can greatly contribute to, or worsen, HF [1].
The proteins involved in redox signaling are protein kinase G (PKG) [2], the small G protein Ras [3], Ca/calmodulin-dependent protein kinase II (CaMKII) [4], protein kinase A (PKA) [5], class II histone deacetylases (HDACs) [6], matrix metalloproteinase (MMP) [7], protein kinase B/Akt [8], the extracellular signal-regulated kinase ½ (ERK1/2) [9], p38 MAP kinase [10], protein kinase C (PKC) [11], NF-kappa B [12], and transcription factors, including activated protein-1 [13].
Cardiomyocyte hypertrophy has been found to be associated with ROS activation of signaling kinases and transcription factors [14]. ROS also promotes post-translational modifications that change the function of specific proteins and signaling pathways, leading to hypertrophic remodeling [14][15]. ROS have been shown to be important in G protein-coupled receptor stimulation by angiotensin II, tumor necrosis factor-α (TNF-α), and α-adrenergic stimulation [14][16][17]. Angiotensin II may participate in myocardial hypertrophy by several intracellular pathways by activating: (1) protein kinase C, (2) c-Jun N-terminal kinase (JNK), (3) extracellular signal-regulated kinase, and (4) ROS formation [16][18]. Though the role of TNF-α in cardiomyocytes is not yet sufficiently known, TNF-α seems to play an autocrine or paracrine role in activating MMPs, which promote hypertrophic changes in the heart [17].
ROS affect different lipid membranes too, including the sarcolemma, mitochondrial membranes, nuclear membrane, and the sarcoplasmic reticulum, in which lipid radicals and lipid hydroperoxide (LPH) form [19][20][21]. As the lipid peroxidation cascade progresses, LPH reacts with fatty acids to form a more stable product, for example—malondialdehyde or 4-hydroxy-2-nonenal [22]. Destabilization of the phospholipid-rich inner mitochondrial membrane by peroxidation results in additional electron leakage and increase in ROS production intensity [20][22].
ROS activate the cardiac Na+/Ca2+ exchanger, which triggers cardiac hypertrophy through the Ca2+-dependent pathway [23] and contributes to Ca2+/calmodulin-dependent protein kinase II activation, leading to increase in cardiomyocyte death and CHF development [24]. The cardiac Na+/H+ exchanger (NHE1) was shown to be activated [25] and sarcolemmal Na+/K+ ATPase was found to be suppressed by ROS [26] and to be implicated in cardiac hypertrophy. It should be emphasized that the elevation of Na+ in cardiomyocytes may contribute to slower cardiac muscle relaxation and arrhythmias [27].
It should also be mentioned that heme oxygenase (HO) (an enzyme that catalyzes heme degradation) has been shown to reduce oxidative stress in cardiomyocytes by catalyzing the carbon monoxide (CO) producing reaction [27][28]. CO has been shown to act as an antioxidant and contribute to the anti-hypertrophic effect [28].
Additionally, ROS induced endothelial damage [29][30] and thrombosis development [31] are stated in the literature to take place in chronic HF development.
2. Enzymes Involved in ROS Production
NADPH oxidases (NOX) 2 and 4 [32], xanthine oxidoreductase (XOR) [33], and nitric oxide synthase (NOS) [34] are the common enzymes that produce ROS in cardiomyocytes (Figure 1).
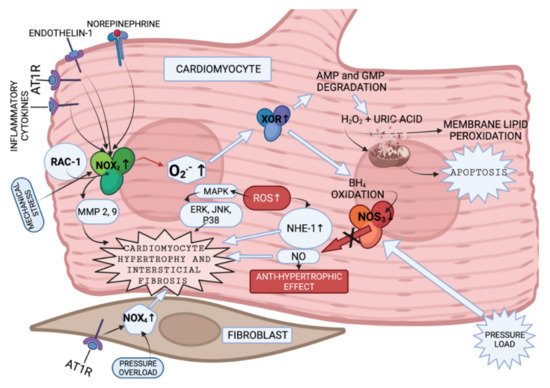
Figure 1. Enzymes involved in ROS production in cardiomyocytes and fibroblasts (created with BioRender.com on 7 February 2022). NOX2 is presented to be activated by endothelin and angiotensin II [35], by cytokines [36] and mechanical stress [37]. Increased NOX2 activation leads to cytoskeletal dysfunction in patients with CHF [38]. It was discovered that superoxide anions, produced by NOX, can oxidize and degrade hydrobiopterin-4 (BH4) leading to NOS uncoupling [39]. Nitric oxide synthase 3 (NOS3) uncoupling was observed in myocardium exposed to chronic pressure load. NOS3 catalyzes nitric oxide (NO) synthesis under physiological conditions. NO has an antihypertrophic effect. However, NOS3 is uncoupled with pressure load, and this, in turn, leads to reduction in tetrahydrobiopterin-4 concentration, increase in ROS production, and, as a consequence, to cardiomyocyte hypertrophy [40]. It was also shown that increase in ROS activates MAPK, leading to increased expression of proteins, such as ERK, JNK and P38, which are related to cardiomyocyte hypertrophy [41] (Figure 1). What is more, NOX-derived ROS may activate XOR [42]. Additionally, angiotensin II-induced signaling and isolated cardiomyocyte hypertrophy are dependent on NOX2 [43]. GTP-binding protein Rac-1 (involved in NOX activation), as described in the literature, is involved in isolated myocyte hypertrophy, induced by endothelin I, phenylephrine, angiotensin II [44] and norepinephrine [45]. AMPK—adenosine monophosphate activated protein kinase; AT1R—angiotensin II receptor; NOX-NADPH oxidase; BH4—dihydrobiopterin-4; NOS—nitric oxide synthase; NO—nitric oxide; MAPK—mitogen-activated protein kinase; XOR—xanthine oxidoreductase; AMP—adenosine monophosphate; GMP—guanosine monophosphate; Rac-1—GTP-binding protein; NHE-1—sodium/hydrogen exchanger-1; ERK—extracellular signal-regulated kinase; JNK-c—Jun N-terminal kinase; p38—a focal point of interactions of the serine/threonine kinases), MMP—matrix metalloproteinase.
Humans have seven NOX with a similar catalytic core, but different regulatory mechanisms [46]. NOX2 and NOX4 are abundantly expressed in cardiomyocytes, endothelium and fibroblasts. Every NOX produces the superoxide anion [39]. NOX2 and NOX4 activity is presented in Figure 1. The data presented in [47][48] implies that NOX4-derived ROS could contribute to overload-induced LV hypertrophy (LVH), and that NOX2 is produced in response to angiotensin II infusion. However, some studies suggest that LVH, as a response to chronic renin-angiotensin-aldosterone system activation, is not associated with NOX2 [49][50]. The enzymes involved in ROS production in cardiomyocytes and fibroblasts, and the pathways that they activate according to the literature [35][36][37][38][39][40][41][42][43][44][45], are presented in summary in Figure 1.
NOX has been shown to be involved in MMP activation in response to angiotensin II [51] and mechanical stretch [49] in the vessels. Experiments with mice and rats have demonstrated the role of NOX2 in the development of interstitial cardiac fibrosis, however, the NOX2-expressing cell type was not established [49][50][52]. NOX4 was shown to be expressed in cardiac fibroblasts [53][54] in animal models. Currently there are no in vivo experiments that could confirm the analogous case in humans.
The other ROS produced enzyme is XOR. XOR is involved in: (1) degradation of the purine nucleotides (AMP and GMP), in which it oxidizes hypoxanthine and xanthine to uric acid and H2O2 [55], (2) reduction of nitrite and nitrate, in which it produces NO and consequently promotes vasodilatation [56], or inflammation [56] and mitochondrial damage (as a result of overproduction) [57]. There are two forms of XOR: xanthine dehydrogenase and xanthine oxidase (XO). XO is involved in H2O2 production [58]. High levels of uric acid are found in patients with HF blood tests [55]; therefore, production of H2O2 is expected to be increased in these patients as well.
NOS catalyze NO production in a reaction where L-arginine is converted to L-citruline [59]. There are three isoforms of NOS. Two isoforms of NOS (endothelial (eNOS) and neuronal (nNOS)) are expressed more intensely in cardiomyocytes [60]. However, iNOS can contribute to contractile damage in CHF as well [61].
Increased ROS production is related both to myosin-activated protein kinase (MAPK) [41] (Figure 1) and adenosine monophosphate activated protein kinase (AMPK) activation [62]. AMPK activation leads to an increase in the antioxidants SOD and catalase (CAT) and uncoupling of protein 2 (UCP2) gene expression, leading to weakened apoptosis and NOX expression decrease [62] (not shown in Figure 1).
Another group of enzymes involved in CHF development through ROS production. is a family of NAD+ dependent class III histone deacetylases called sirtuins (SIRT) [63]. There are seven members of this enzyme group in different cell departments. SIRT3 is found in the mitochondria [64] and is involved in ATP production and ROS detoxication [65][66]. Some studies have found that SIRT3 is involved in cardiac muscle hypertrophy and fibrosis, leading eventually to CHF development [67][68]. Additionally, some studies have found that SIRT3 is involved in oxidative stress-mediated cell death in cardiomyocytes through protein Ku70 deacetylation, leading to deacetylated Ku70 interaction with the apoptosis regulator bcl-2-like protein4 (Bax) [69]. However, other studies have highlighted several mechanisms through which SIRT3 exerts a cardioprotective effect: (1) SIRT3 activates the antioxidant enzyme superoxide dismutase (SOD) [70], and (2) activates isocitrate dehydrogenase 2 (IDH2) by deacetylation. IDH2 uses NADP+ for reduction. SIRT3, in this way, increases NADPH levels, and increases glutathione (GSH) levels, thereby, inhibiting ROS production [71].
It can be concluded that several redox-signaling pathways may be modulated by ROS-producing enzymes, leading to cardiomyocyte hypertrophy, interstitial fibrosis and apoptosis. The value of NOX seems to be far more important in comparison with the other enzymes in that its activation can be triggered by both neuroendocrine factors and pressure overload, or by inflammatory cytokines. NOS play an important role in redox alterations in CHF development, both with substrates and cofactors. Sirtuins are involved in the enzyme-antioxidant activation that protects the heart from hypertrophy. NOX-, XOR-, NOS- and SIRT-mediated pathways could, therefore, be potential treatment targets for CHF development suppression.
3. Mitochondria in ROS Production and Enzyme-Antioxidants
The most abundant source of ROS in cells is the mitochondrial electron transport chain (ETC). A total of 0.2–2% of the electrons in the ETC leak out of the chain and interact with oxygen to produce superoxide or hydrogen peroxide [24]. Additionally, H2O2 is produced in the reaction, catalyzed by SOD1. After it has diffused from mitochondria, H2O2 is involved in physiological and pathological pathways (damaging proteins and lipids) [24]. H2O2 damages mitochondrial DNA, interferes with the Krebs cycle, ATP production, and fatty acid metabolism [72] and can trigger the opening of ion channels or the inner membrane anion channel inside the mitochondria, leading to cell death [73]. Proton leakage in the mitochondria consists of: (1) basal leakage (not regulated and related to the inner mitochondrial membrane’s lipid bilayer and the adenine nucleotide translocase (ANT)), and (2) inducible leakage (regulated and catalyzed or suppressed by uncoupling proteins and ANT) [24]. UCP2 is involved in cardiovascular disease; therefore, drugs targeting UCP expression or activity might be a potential treatment option. Hypoxia is suggested to further increase ROS production in the mitochondria [24]. ETC complexes III (CIII), and especially I (CI), are found to be the main sites of ROS production [74]. Therefore, the regulation of ROS production in these complexes may yield significant results.
Mitochondrial ROS are also involved in different cell signaling pathways, involving apoptosis [75], autophagy [76], and necrosis [77].
Other ROS sources in the mitochondria are the enzymes monoamine oxidase A and B, both located within the outer mitochondrial membrane (OMM). They catalyze the oxidative deamination of neurotransmitters and biogenic amines, leading to H2O2 production [78]. Monoamine oxidase A (MAO-A) is specific to cardiomyocytes [79]. It was discovered that MAO-A-dependent ROS formation may impair autophagy, leading to the accumulation of autophagosomes and mitochondrial fusion, resulting in microtubule-associated protein light chain 3-phosphatidylethanolamine conjugate (recruited to autophagosomal membranes) formation, and autophagy receptor (p62) and ubiquitylated protein accumulation, causing cardiomyocyte death and CHF. Both MAO-A derived H2O2 and aldehydes are able to directly target mitochondrial function [80]. Additionally, MAO-A-generated ROS has been shown to inhibit sphingosine kinase, which leads to ceramide accumulation and, thereby, to cardiomyocyte apoptosis [81]. Contractile proteins (actin and tropomyosin) are also affected [82].
Excess of H2O2 in the cell is eliminated by glutathione peroxidase (GPX) and peroxiredoxin (PRX). Both of these require GSH and thioredoxin for regeneration [83], which requires NADPH as a cofactor [83][84]. Nicotinamide nucleotide transhydrogenase, NADP+-dependent isocitrate dehydrogenase (IDH) and malic enzyme are involved in NADP+ regeneration to NADPH [85]. These require Krebs cycle products—NADH, malate and isocitrate. With IDH being the most important for NADPH regeneration [86], the Krebs cycle is necessary for the antioxidant capacity within mitochondria [87] as well as in the cytosol [88]. Aldehyde dehydrogenase (ALDH2) is another mitochondrial enzyme involved in antioxidant activity and participates in the detoxication of lipid peroxidation products [89]. Moreover, ROS through intermediate links, activate AMPK, leading to an increase in antioxidant enzyme gene expression (SOD, CAT and (UCP2)) [62].
In sum, the mitochondria are an important ROS source in cardiomyocytes. The amount of ROS produced by mitochondria depends on the supply of oxygen to the cell and activity of the enzymes that produce ROS (especially SOD1 and MAO-A). Cardiomyocyte cytosol antioxidant GPX and PRX regeneration also depends on Krebs cycle action. AMPK activation is important for antioxidant enzyme function.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10030602
References
- Weissman, D.; Maack, C. Redox signaling in heart failure and therapeutic implications. Free Radic. Biol. Med. 2021, 171, 345–364.
- Burgoyne, J.R.; Madhani, M.; Cuello, F.; Charles, R.L.; Brennan, J.P.; Schröder, E. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 2007, 317, 1393–1397.
- Kuster, G.M.; Siwik, D.A.; Pimentel, D.R.; Colucci, W.S. Role of Reversible, Thioredoxin-Sensitive Oxidative Protein Modifications in Cardiac Myocytes. Antioxid. Redox Signal. 2006, 8, 2153–2159.
- Erickson, J.R.; Joiner, M.-L.A.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A Dynamic Pathway for Calcium-Independent Activation of CaMKII by Methionine Oxidation. Cell 2008, 133, 462–474.
- Brennan, J.P.; Bardswell, S.C.; Burgoyne, J.R.; Fuller, W.; Schröder, E.; Wait, R. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J. Biol. Chem. 2006, 281, 21827–21836.
- Ago, T.; Liu, T.; Zhai, P.; Chen, W.; Li, H.; Molkentin, J.; Vatner, S.F.; Sadoshima, J. A Redox-Dependent Pathway for Regulating Class II HDACs and Cardiac Hypertrophy. Cell 2008, 133, 978–993.
- Sood, H.S.; Cox, M.J.; Tyagi, S.C. Generation of Nitrotyrosine Precedes Activation of Metalloproteinase in Myocardium of Hyperhomocysteinemic Rats. Antioxid. Redox Signal. 2002, 4, 799–804.
- Yasukawa, T.; Tokunaga, E.; Ota, H.; Sugita, H.; Martyn, J.A.J.; Kaneki, M. S-Nitrosylation-dependent Inactivation of Akt/Protein Kinase B in Insulin Resistance. J. Biol. Chem. 2005, 280, 7511–7518.
- Kehat, I.; Molkentin, J.D. Extracellular signal-regulated kinase 1/2 (ERK1/2) signaling in cardiac hypertrophy. Ann. N. Y. Acad. Sci. 2010, 1188, 96–102.
- Meijles, D.N.; Cull, J.J.; Markou, T.; Cooper, S.T.; Haines, Z.H.; Fuller, S.J.; O’Gara, P.; Sheppard, M.N.; Harding, S.; Sugden, P.H.; et al. Redox Regulation of Cardiac ASK1 (Apoptosis Signal-Regulating Kinase 1) Controls p38-MAPK (Mitogen-Activated Protein Kinase) and Orchestrates Cardiac Remodeling to Hypertension. Hypertension 2020, 76, 1208–1218.
- Newton, A.C.; Antal, C.E.; Steinberg, S.F. Protein kinase C mechanisms that contribute to cardiac remodelling. Clin. Sci. 2016, 130, 1499–1510.
- Gao, G.; Dudley, S.C. Redox regulation, NF-kappaB, and atrial fibrillation. Antioxid. Redox Signal 2009, 11, 2265–2277.
- Sasaki, H.; Galang, N.; Maulik, N. Redox regulation of NF-kappaB and AP-1 in ischemic reperfused heart. Antioxid. Redox Signal 1999, 1, 317–324.
- Takimoto, E.; Kass, D.A. Role of Oxidative Stress in Cardiac Hypertrophy and Remodeling. Hypertension 2007, 49, 241–248.
- Ho, E.; Karimi Galougahi, K.; Liu, C.-C.; Bhindi, R.; Figtree, G.A. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biol. 2013, 1, 483–491.
- Nakamura, K.; Fushimi, K.; Kouchi, H.; Mihara, K.; Miyazaki, M.; Ohe, T.; Namba, M. Inhibitory Effects of Antioxidants on Neonatal Rat Cardiac Myocyte Hypertrophy Induced by Tumor Necrosis Factor-α and Angiotensin II. Circulation 1998, 98, 794–799.
- Yokoyama, T.; Nakano, M.; Bednarczyk, J.L.; McIntyre, B.W.; Entman, M.; Mann, U.L. Tumor Necrosis Factor-α Provokes a Hypertrophic Growth Response in Adult Cardiac Myocytes. Circulation 1997, 95, 1247–1252.
- Yamazaki, T.; Komuro, I.; Zou, Y.; Kudoh, S.; Shiojima, I.; Hiroi, Y. Norepinephrine induces the raf-1 kinase/mitogen-activated protein kinase cascade through both alpha 1- and beta-adrenoceptors. Circulation 1997, 95, 1260–1268.
- Christiansen, L.B.; Dela, F.; Koch, J.; Hansen, C.N.; Leifsson, P.S.; Yokota, T. Impaired cardiac mitochondrial oxidative phosphorylation and enhanced mitochondrial oxidative stress in feline hypertrophic cardiomyopathy. Am. J. Physiol. Circ. Physiol. 2015, 308, H1237–H1247.
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495.
- Wijnker, P.J.; Sequeira, V.; Kuster, D.W.; Van Der Velden, J. Hypertrophic Cardiomyopathy: A Vicious Cycle Triggered by Sarcomere Mutations and Secondary Disease Hits. Antioxid. Redox Signal. 2019, 31, 318–358.
- Del Rio, D.; Stewart, A.J.; Pellegrini, N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 316–328.
- Horacio, E.C.; Cingolani, H.E.; Perez, N.G.; Aiello, E.A.; Ennis, I.L.; Garciarena, C.D.; Villa-Abrille, M.C.; Dulce, R.A.; Caldiz, C.I.; Yeves, A.M.; et al. Early signals after stretch leading to cardiac hypertrophy. Key role of NHE-1. Front. Biosci. 2008, 13, 7096–7114.
- Palomeque, J.; Rueda, O.V.; Sapia, L.; Valverde, C.A.; Salas, M.; Petroff, M.V.; Mattiazzi, A. Angiotensin II–Induced Oxidative Stress Resets the Ca2+ Dependence of Ca2+–Calmodulin Protein Kinase II and Promotes a Death Pathway Conserved Across Different Species. Circ. Res. 2009, 105, 1204–1212.
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15.
- Marck, P.; Pessoa, M.; Xu, Y.; Kutz, L.; Collins, D.; Yan, Y.; King, C.; Wang, X.; Duan, Q.; Cai, L.; et al. Cardiac Oxidative Signaling and Physiological Hypertrophy in the Na/K-ATPase α1s/sα2s/s Mouse Model of High Affinity for Cardiotonic Steroids. Int. J. Mol. Sci. 2021, 22, 3462.
- Pieske, B.; Maier, L.S.; PiacentinoIII, V.; Weisser, J.; Hasenfuss, G.; Houser, S. Rate Dependence of i and Contractility in Nonfailing and Failing Human Myocardium. Circulation 2002, 106, 447–453.
- Liu, Y.; Wang, H.; Wang, J.; Wei, B.; Zhang, X.; Zhang, M.; Cao, D.; Dai, J.; Wang, Z.; Nyirimigabo, E.; et al. A positive feedback regulation of Heme oxygenase 1 by CELF1 in cardiac myoblast cells. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 209–218.
- AlHayaza, R.; Haque, E.; Karbasiafshar, C.; Sellke, F.W.; Abid, M.R. The Relationship Between Reactive Oxygen Species and Endothelial Cell Metabolism. Front. Chem. 2020, 8, 592688.
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19.
- Cimmino, G.; Cirillo, P.; Ragni, M.; Conte, S.; Uccello, G.; Golino, P. Reactive oxygen species induce a procoagulant state in endothelial cells by inhibiting tissue factor pathway inhibitor. J. Thromb. Thrombolysis 2015, 40, 186–192.
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH Oxidases in Heart Failure: Poachers or Gamekeepers? Antioxid. Redox Signal 2013, 18, 1024.
- Ekelund, U.E.G.; Harrison, R.W.; Shokek, O.; Thakkar, R.N.; Tunin, R.S.; Senzaki, H.; Kass, D.A.; Marbaán, E.; Hare, J.M. Intravenous Allopurinol Decreases Myocardial Oxygen Consumption and Increases Mechanical Efficiency in Dogs with Pacing-Induced Heart Failure. Circ. Res. 1999, 85, 437–445.
- Jankovic, A.; Korac, A.; Buzadzic, B.; Stancic, A.; Otasevic, V.; Ferdinandy, P.; Daiber, A.; Korac, B. Targeting the NO/superoxide ratio in adipose tissue: Relevance to obesity and diabetes management. J. Cereb. Blood Flow Metab. 2016, 174, 1570–1590.
- Looi, Y.H.; Grieve, D.J.; Siva, A.; Walker, S.J.; Anilkumar, N.; Cave, A.C.; Marber, M.; Monaghan, M.J.; Shah, A.M. Involvement of Nox2 NADPH Oxidase in Adverse Cardiac Remodeling After Myocardial Infarction. Hypertension 2008, 51, 319–325.
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, Physiology, and Pathophysiology of NADPH Oxidases in the Cardiovascular System. Circ. Res. 2012, 110, 1364–1390.
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS Signaling: Rapid Mechano-Chemo Transduction in Heart. Science 2011, 333, 1440–1445.
- Izu, L.T.; Kohl, P.; Boyden, P.A.; Miura, M.; Banyasz, T.; Chiamvimonvat, N. Mechano-Electric and Mechano-Chemo-Transduction in Cardiomyocytes. J. Physiol. 2020, 598, 1285.
- Murdoch, C.E.; Zhang, M.; Cave, A.C.; Shah, A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006, 71, 208–215.
- Takimoto, E.; Champion, H.C.; Li, M.; Ren, S.; Rodriguez, E.R.; Tavazzi, B.; Lazzarino, G.; Paolocci, N.; Gabrielson, K.L.; Wang, Y.; et al. Oxidant stress from nitric oxide synthase–3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J. Clin. Investig. 2005, 115, 1221–1231.
- Cheng, D.; Chen, L.; Tu, W.; Wang, H.; Wang, Q.; Meng, L. Protective effects of valsartan administration on doxorubicin-induced myocardial injury in rats and the role of oxidative stress and NOX2/NOX4 signaling. Mol. Med. Rep. 2020, 22, 4151–4162.
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Circ. Physiol. 2003, 285, H2290–H2297.
- Nakagami, H.; Takemoto, M.; Liao, J.K. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2003, 35, 851–859.
- Higuchi, Y.; Otsu, K.; Nishida, K.; Hirotani, S.; Nakayama, H.; Yamaguchi, O. The Small GTP-binding Protein Rac1 Induces Cardiac Myocyte Hypertrophy through the Activation of Apoptosis Signal-regulating Kinase 1 and Nuclear Factor-κB*. J. Biol. Chem. 2003, 278, 20770–20777.
- Xiao, L.; Pimental, D.R.; Amin, J.K.; Singh, K.; Sawyer, D.B.; Colucci, W.S. MEK1/2-ERK1/2 mediates alpha1-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J. Mol. Cell Cardiol. 2001, 33, 779–787.
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106.
- Byrne, J.A.; Grieve, D.J.; Bendall, J.K.; Li, J.-M.; Gove, C.; Lambeth, J.D.; Cave, A.C.; Shah, A.M. Contrasting Roles of NADPH Oxidase Isoforms in Pressure-Overload Versus Angiotensin II–Induced Cardiac Hypertrophy. Circ. Res. 2003, 93, 802–805.
- Bendall, J.K.; Cave, A.C.; Heymes, C.; Gall, N.; Shah, A.M. Pivotal Role of a gp91 phox -Containing NADPH Oxidase in Angiotensin II-Induced Cardiac Hypertrophy in Mice. Circulation 2002, 105, 293–296.
- Grote, K.; Flach, I.; Luchtefeld, M.; Akin, E.; Holland, S.M.; Drexler, H.; Schieffer, B. Mechanical Stretch Enhances mRNA Expression and Proenzyme Release of Matrix Metalloproteinase-2 (MMP-2) via NAD(P)H Oxidase–Derived Reactive Oxygen Species. Circ. Res. 2003, 92, e80–e86.
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-induced inflammation in the rat heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781.
- Luchtefeld, M.; Grote, K.; Grothusen, C.; Bley, S.; Bandlow, N.; Selle, T.; Strüber, M.; Haverich, A.; Bavendiek, U.; Drexler, H.; et al. Angiotensin II induces MMP-2 in a p47phox-dependent manner. Biochem. Biophys. Res. Commun. 2005, 328, 183–188.
- Johar, S.; Cave, A.C.; Narayanapanicker, A.; Grieve, D.J.; Shah, A.M. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. 2006, 20, 1546–1548.
- Colston, J.T.; De La Rosa, S.D.; Strader, J.R.; Anderson, M.A.; Freeman, G.L. H2O2 activates Nox4 through PLA2-dependent arachidonic acid production in adult cardiac fibroblasts. FEBS Lett. 2005, 579, 2533–2540.
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907.
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797.
- Harrison, R. Physiological Roles of Xanthine Oxidoreductase. Drug Metab. Rev. 2004, 36, 363–375.
- Gladden, J.D.; Zelickson, B.R.; Wei, C.-C.; Ulasova, E.; Zheng, J.; Ahmed, M.I.; Chen, Y.; Bamman, M.; Ballinger, S.; Darley-Usmar, V.; et al. Novel insights into interactions between mitochondria and xanthine oxidase in acute cardiac volume overload. Free Radic. Biol. Med. 2011, 51, 1975–1984.
- Watanabe, K.; Watanabe, T.; Otaki, Y.; Shishido, T.; Murase, T.; Nakamura, T.; Kato, S.; Tamura, H.; Nishiyama, S.; Takahashi, H.; et al. Impact of plasma xanthine oxidoreductase activity in patients with heart failure with preserved ejection fraction. ESC Heart Fail. 2020, 7, 1735–1743.
- Semenza, G.L. Hypoxia-Inducible Factor 1 and Cardiovascular Disease. Annu. Rev. Physiol. 2014, 76, 39–56.
- Massion, P.B.; Balligand, J.-L. Relevance of nitric oxide for myocardial remodeling. Curr. Heart Fail. Rep. 2007, 4, 18–25.
- Soskić, S.S.; Dobutović, B.D.; Sudar, E.M.; Obradović, M.M.; Nikolić, D.M.; Djordjevic, J.D.; Radak, D.J.; Mikhailidis, D.P.; Isenović, E.R. Regulation of Inducible Nitric Oxide Synthase (iNOS) and its Potential Role in Insulin Resistance, Diabetes and Heart Failure. Open Cardiovasc. Med. J. 2011, 5, 153–163.
- Marino, A.; Hausenloy, D.J.; Andreadou, I.; Horman, S.; Bertrand, L.; Beauloye, C. AMP-activated protein kinase: A remarkable contributor to preserve a healthy heart against ROS injury. Free Radic. Biol. Med. 2021, 166, 238–254.
- North, B.J.; Sinclair, D.A. The Intersection Between Aging and Cardiovascular Disease. Circ. Res. 2012, 110, 1097–1108.
- Finkel, T.; Deng, C.-X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591.
- Wang, T.; Cao, Y.; Zheng, Q.; Tu, J.; Zhou, W.; He, J.; Zhong, J.; Chen, Y.; Wang, J.; Cai, R.; et al. SENP1-Sirt3 Signaling Controls Mitochondrial Protein Acetylation and Metabolism. Mol. Cell 2019, 75, 823–834.e5.
- Koentges, C.; Cimolai, M.C.; Pfeil, K.; Wolf, D.; Marchini, T.O.; Tarkhnishvili, A.; Hoffmann, M.M.; Odening, K.E.; Diehl, P.; Mühlen, C.V.Z.; et al. Impaired SIRT3 activity mediates cardiac dysfunction in endotoxemia by calpain-dependent disruption of ATP synthesis. J. Mol. Cell. Cardiol. 2019, 133, 138–147.
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771.
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783.
- Luo, K.; Huang, W.; Tang, S. Sirt3 enhances glioma cell viability by stabilizing Ku70–BAX interaction. OncoTargets Ther. 2018, 11, 7559–7567.
- Tseng, A.H.; Shieh, S.-S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234.
- Bergaggio, E.; Riganti, C.; Garaffo, G.; Vitale, N.; Mereu, E.; Bandini, C.; Pellegrino, E.; Pullano, V.; Omedè, P.; Todoerti, K.; et al. IDH2 inhibition enhances proteasome inhibitor responsiveness in hematological malignancies. Blood 2019, 133, 156–167.
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13.
- Anderson, E.J.; Rodriguez, E.; Anderson, C.A.; Thayne, K.; Chitwood, W.R.; Kypson, A.P. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am. J. Physiol. Circ. Physiol. 2011, 300, H118–H124.
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472.
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180.
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026.
- Morgan, M.J.; Liu, Z.G. Reactive oxygen species in TNFalpha-induced signaling and cell death. Mol. Cells 2010, 30, 1–12.
- Anderson, E.J.; Efird, J.T.; Davies, S.W.; O’Neal, W.T.; Darden, T.M.; Thayne, K.A.; Katunga, L.A.; Kindell, L.C.; Ferguson, T.B.; Anderson, C.A.; et al. Monoamine Oxidase is a Major Determinant of Redox Balance in Human Atrial Myocardium and is Associated with Postoperative Atrial Fibrillation. J. Am. Heart Assoc. 2014, 3, e000713.
- Santin, Y.; Sicard, P.; Vigneron, F.; Guilbeau-Frugier, C.; Dutaur, M.; Lairez, O.; Couderc, B.; Manni, D.; Korolchuk, V.I.; Lezoualc’H, F.; et al. Oxidative Stress by Monoamine Oxidase-A Impairs Transcription Factor EB Activation and Autophagosome Clearance, Leading to Cardiomyocyte Necrosis and Heart Failure. Antioxid. Redox Signal. 2016, 25, 10–27.
- Deshwal, S.; Di Sante, M.; Di Lisa, F.; Kaludercic, N. Emerging role of monoamine oxidase as a therapeutic target for cardiovascular disease. Curr. Opin. Pharmacol. 2017, 33, 64–69.
- Pchejetski, D.; Kunduzova, O.; Dayon, A.; Calise, D.; Seguelas, M.-H.; Leducq, N.; Seif, I.; Parini, A.; Cuvillier, O. Oxidative Stress–Dependent Sphingosine Kinase-1 Inhibition Mediates Monoamine Oxidase A–Associated Cardiac Cell Apoptosis. Circ. Res. 2007, 100, 41–49.
- Menazza, S.; Blaauw, B.; Tiepolo, T.; Toniolo, L.; Braghetta, P.; Spolaore, B.; Reggiani, C.; Di Lisa, F.; Bonaldo, P.; Canton, M. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum. Mol. Genet. 2010, 19, 4207–4215.
- Molavian, H.; Tonekaboni, A.M.; Kohandel, M.; Sivaloganathan, S. The Synergetic Coupling among the Cellular Antioxidants Glutathione Peroxidase/Peroxiredoxin and Other Antioxidants and its Effect on the Concentration of H2O2. Sci. Rep. 2015, 5, srep13620.
- Aon, M.A.; Stanley, B.A.; Sivakumaran, V.; Kembro, J.M.; O’Rourke, B.; Paolocci, N.; Cortassa, S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: An experimental-computational study. J. Gen. Physiol. 2012, 139, 479–491.
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal 2008, 10, 179–206.
- Nickel, A.G.; Von Hardenberg, A.; Hohl, M.; Löffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.-C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015, 22, 472–484.
- Kohlhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Böhm, M.; O’Rourke, B.; Maack, C. Elevated Cytosolic Na+ Increases Mitochondrial Formation of Reactive Oxygen Species in Failing Cardiac Myocytes. Circulation 2010, 121, 1606–1613.
- Schulz, E.; Wenzel, P.; Münzel, T.; Daiber, A. Mitochondrial Redox Signaling: Interaction of Mitochondrial Reactive Oxygen Species with Other Sources of Oxidative Stress. Antioxid. Redox Signal. 2014, 20, 308–324.
- Wenzel, P.; Müller, J.; Zurmeyer, S.; Schuhmacher, S.; Schulz, E.; Oelze, M.; Pautz, A.; Kawamoto, T.; Wojnowski, L.; Kleinert, H.; et al. ALDH-2 deficiency increases cardiovascular oxidative stress—Evidence for indirect antioxidative properties. Biochem. Biophys. Res. Commun. 2008, 367, 137–143.
This entry is offline, you can click here to edit this entry!