Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Respiratory System
Over the past decades, a better understanding of the genetic and molecular alterations underlying several respiratory diseases has encouraged the development of new therapeutic strategies. Gene therapy offers new therapeutic alternatives for inherited and acquired diseases by delivering exogenous genetic materials into cells or tissues to restore physiological protein expression and/or activity.
- gene therapy
- treatment
- respiratory disease
- gene editing
- AAV
- Lentivirus
- Nanoparticles
- gene transfer
- Viral Vector Systems
1. Viral Vectors
1.1. Retroviral and Lentiviral Vectors
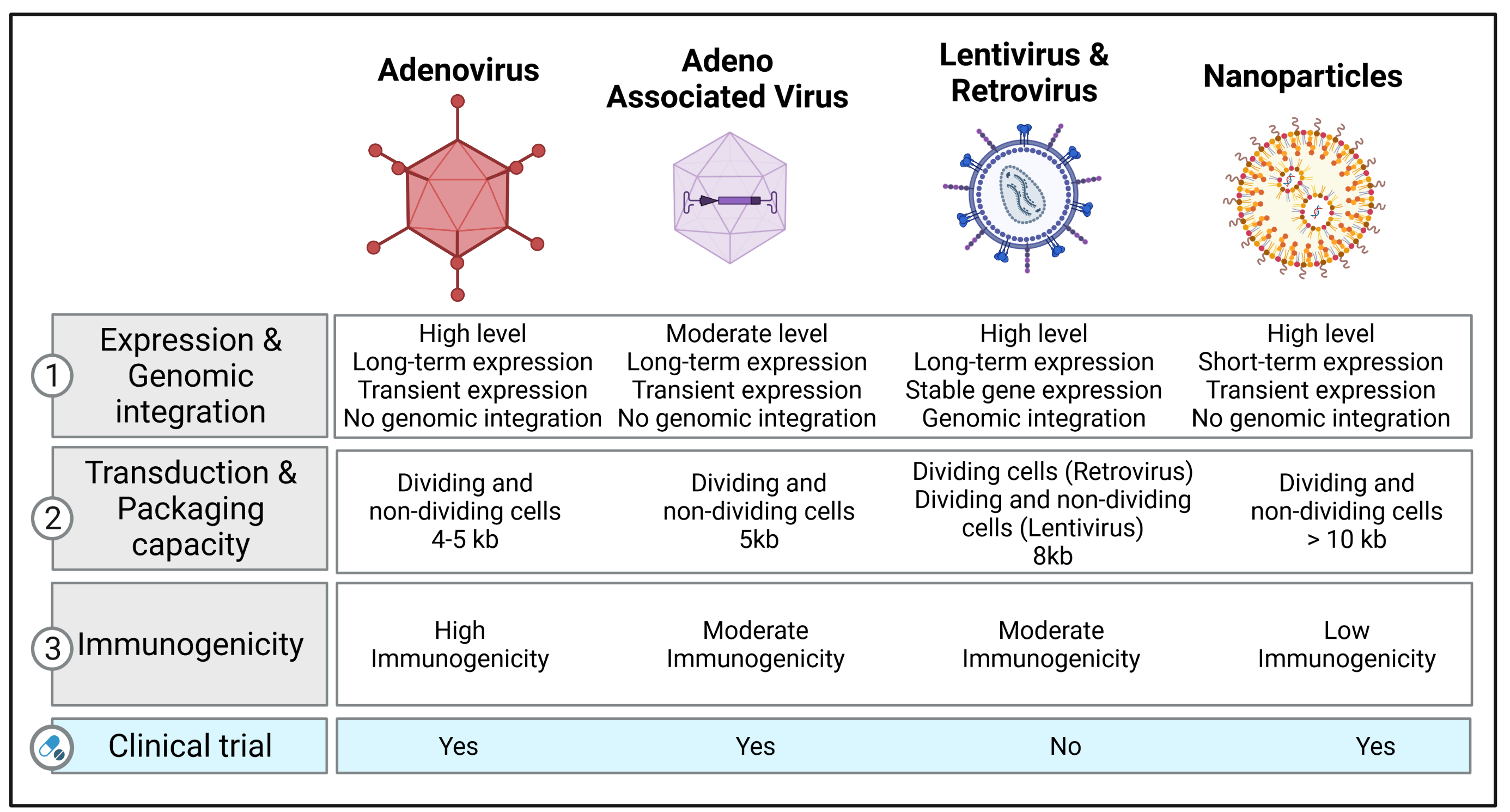
Retrovirus and lentivirus genomes are characterized by two long terminal repeats at either end of the genome and a packaging sequence. A transgene cassette up to 8 kb can be inserted in the space by removing native genes, such as GAG, POL, and ENV. The RNA genome is converted into double-stranded DNA that can randomly integrate into the host’s chromosomal DNA during the viral life cycle. As a result, retroviral and lentiviral vectors have been investigated in proliferating cells, with attempts to achieve stable transduction by transmitting the therapeutic gene to daughter cells [7]. Consequently, the use of retroviral vectors for in vivo applications is not recommended, as lung cells only show low cell proliferation rates [8].
Lentiviral vectors can infect post-mitotic cells [9] with higher transduction rates. Lentiviral vectors have been broadly used in clinical trials for treating Parkinson’s disease and immunodeficiency disorders [10]. Moreover, lentiviral vectors have been approved for treating certain lymphomas and leukemias, which mainly involve the ex vivo treatment of hematopoietic stem cells [11]. Recently, several preclinical studies have shown encouraging data with lentiviral vectors regarding safety [12,13]; further clinical studies are needed to evaluate the long-term safety and efficacy. The use of lentiviral vectors for lung gene delivery is limited due to the barriers in the lung and the low localized expression of cellular receptors on the apical surface of the lung epithelial cells [14]. This can be overcome, in part, by using pseudotyped lentiviral vectors from diverse origins, such as Ebola, Sendai virus, or influenza. Recent studies have found multiple pseudotyped lentiviral vectors that can achieve robust apical transduction in epithelial cells in vivo, such as G64-pseudotyped or Sendai virus-pseudotyped lentiviral vectors [13,15,16,17]. However, the potential integration of the transgene into the host genome limits their use in clinical settings. The risk of random integration could also potentially activate proto-oncogenes or inactivate tumor suppressor genes [18].
1.2. Adenovirus Vectors
Recombinant adenovirus (Ad) vectors are characterized by a natural tropism for airway epithelial cells in the respiratory tract, making them efficient vehicles for lung gene transfer. Ad vectors consist of a double-stranded genome. E1 gene deletion allows preventing vector replication and provides greater packaging space for the transgene [19]. These vectors can transduce proliferating and non-dividing cells. They are relatively easy to produce and purify at a large scale. Due to the highly immunogenic nature of Ad vectors, host immunity may limit the gene expression to 2–3 weeks [20]. For example, Ad-mediated gene transfer can cause an immune response and apoptosis in endothelial cells in organ-cultured pulmonary arteries, which leads to low levels of gene transfer [21]. The genetic material of Ad vectors is not integrated into the host’s cells genome, but rather remains episomal in the nucleus. Thus, the long-term episomal persistence of transgenes depends on the proliferative state of target cells. However, proliferating cells dilute the transgene expression, since only daughter cells express the exogenous transgene. As previously mentioned, Ad vectors induce humoral and cellular immune responses [22]. They are, therefore, unsuitable for repeated administration. Similar to retroviral and lentiviral vectors, the receptors for entry and internalization are mainly located in the basolateral surface. Since epithelial cells have tight junctions, vector–receptor interactions may be limited. Despite these limitations, there is an extensive safety record for lung gene therapy [8]. Second-generation Ad vectors have been developed to minimize immunogenicity and reduce the production of viral antigens by deleting the E2 and E4 viral genes [20]. These new vectors also partially reduce the vector-induced inflammatory response. More recently, “gutted/helper-dependent” adenovirus (HD-Ad) vectors have been developed by removing viral genes, with a reduced immunogenicity of Ad vectors and a large packaging capacity of up to 36 kb [23]. HD-Ad vectors expressing CFTR can efficiently transduce and restore CFTR activity in vitro and in vivo [24].
1.3. Adeno-Associated Viruses (AAV) and Their Vectors
AAV are nonpathogenic, single-stranded parvoviruses and do not cause any diseases. The Rep and Cap genes are bound by two inverted terminal repeats in wild-type AAV. In recombinant AAV (rAAV) vectors, the Rep and Cap genes are replaced by the cDNA encoding the transgene of interest. Recombinant AAV vectors are not integrated into the host genome. The production of AAV requires the co-transfection of the AAV construct with a plasmid containing a helper. AAV can also be produced by infecting packaging cell lines with a recombinant helper containing the rAAV genome. AAV vectors have demonstrated better transduction efficiency and long-term transgene expression in quiescent and dividing cells.
Contrary to Ad vectors, AAV vectors do not induce a strong immune response. AAV have a broad host range and have been commonly used to transfer genes to the airway epithelium, alveolar epithelium, pulmonary vascular endothelium, and pleural mesothelioma [25]. Initially, only two serotypes of the AAV vectors, 2 and 5, were available. More than 100 serotypes have since been developed to enhance cellular and tissue tropism, expand their therapeutic applications to several diseases, and limit off-target effects. However, the size of the expression cassette is limited at 5 kb, which may narrow their application to “small” transgenes. Recently, the application of a smaller Cas variant or dual AAV vectors may provide a solution to the packaging size limitation. Indeed, the strategy of dual AAV vectors has been shown to reach effective transduction levels in pig and mice lungs [26,27]. The packaging capacity of AAV vectors can be increased by creating a parvovirus chimera, with the rAAV genome packaged in the capsid of another parvovirus, such as human bocavirus (HBoV) or gorilla bocavirus (GBoV). Recent studies have confirmed the transduction efficacy of rAAV/HBoV and rAAV/GBoV in primary human airway epithelial cells, lung organoids, and ferret lungs [28,29,30]. Currently, the use of AAV vectors is among the most frequently used strategies for gene therapy. While they have shown promising results in patients, one major concern is that much of the human population has already been exposed to various AAV serotypes, and as a result, they may already exhibit neutralizing antibodies that can significantly impair future gene transfer applications [31]. New recombinant AAV (rAAV), such as site-mutated AAV, are now being developed and optimized to evade the immune response associated with pre-existing neutralizing antibodies, while improving the AAV tropism and increasing transduction efficiency [32,33,34]. For example, site-directed mutagenesis (tyrosine to phenylalanine) has been used to impair the ubiquitination of surface-exposed tyrosine residues, which significantly decreased proteasome-mediated degradation and improved vector transduction [32,35]. Previous studies from Petrs-Silva et al. reported that the delivery of mutated AAV2, AAV8, and AAV9 in mouse retina enhanced transduction efficiency using mutated AAV2, AAV8, and AAV9 [36]. Similarly, Martini et al. investigated whether tyrosine mutations improve the gene transfer efficiency of AAV8 in the lung after intratracheal delivery. Consistently, they found that tyrosine-mutant AAV8 vectors enhanced the transduction efficiency in the lung without inducing any significant changes in lung mechanics, morphometry, or inflammatory response [37]. Another study assessed the effects of single-strand or self-complementary recombinants of AAV vectors containing single or multiple tyrosine-to-phenylalanine (Y-F) mutations in capsid surface-exposed residues on AAV serotypes 2, 8, or 9 [38]. The authors identified six rAAV vectors that showed higher transduction efficacy in CF bronchial epithelial cells by overcoming the intracellular trafficking and second-strand DNA synthesis limitations [38].
1.4. Other Viral Vectors
Many other viruses, such as polyomaviruses, Vaccinia virus, baculovirus, and Sendai virus, have been used for lung gene therapy. Polyomaviruses and John Cunningham virus can provide sustained transgene expression by integrating into the host genome in dividing and non-dividing cells. These vectors can be produced and purified in high titers; however, they showed a limited packaging capacity of approximately 2.5–5 kb and potential for random integration [39]. Vaccinia virus (VV) vectors allow the insertion of large DNA fragments up to 25 kb in size. They represent a promising tool for lung gene transfer as they can infect most cells. Conversely, VV vectors may induce cytopathic effects and immune responses. They may also have limited efficacy due to neutralizing antibodies associated with infections. These properties have been further investigated for treating lung cancer [40]. Baculovirus expression vectors (BEV) have been used for recombinant protein expression and can carry up to 38 kb. BEV cannot replicate in mammalian cells and have no pre-existing BEV immunity in humans. The short-term transgene expression, the entry process through the basolateral membrane, and rapid virus inactivation by serum complements represent major challenges that should be considered for vector optimization [41]. Finally, Sendai virus (SeV)-derived vectors have shown a transduction efficiency in mice in vivo that is 3–4 logs higher than Ad5 or plasmid/liposomes [42]. Since transgene expression is transient, repeated administration leads to diminished gene expression. Pseudotyping SeV envelope proteins onto lentivirus can overcome this limitation [13].
2. Non-Viral Vectors
Different types of non-viral vectors can be used to provide the therapeutic genetic material as naked DNA or RNA (messenger RNA (mRNA), short double-stranded RNA, including small interfering RNA (iRNA) and microRNA (miRNA) mimics, or modified mRNA (modRNA)) or complexed with other macromolecules to enhance the efficiency of cell entry. RNA-based gene therapy has successfully overcome the limitations previously identified with viral vectors such as adenovirus, AAV, retroviral, and lentiviral vectors, including (1) the size capacity of the expression cassette, (2) immunogenicity, and (3) insertional mutagenesis. The advantages of non-viral systems include the ease of vector production, greater expression cassette size, and relatively minimal biosafety risks. Importantly, RNA does not require nuclear localization or transcription, whereas the transfer of DNA cargo requires translocation into the nucleus. An additional benefit is the negligible risk of genomic integration of the delivered sequence. Even though these vectors have significant advantages, they only displayed short-term expression, limited tropism, and moderate efficiency in vivo [43].
2.1. Nanoparticle-Based Therapeutics
Recent advances in nanoparticle-based therapeutics have led to new delivery systems that can deliver siRNAs, exogenous DNA, and mRNA to cells. Liposomes combined with plasmids have been used to enhance the internalization of the genetic material into the cytosol via endocytosis. Nanoparticles consist of a nucleic acid complex with other materials, such as lipid, polymers, peptides, and polysaccharides [44]. Solid lipid nanoparticles that can remain solid at physiological temperatures have been employed because they stay protected from nucleic acid degradation caused by the nuclease. Despite the risk of cytotoxicity, synthetic polymers, such as polyethyleneimine (PEI) or polyethylene glycol (PEG), are commonly used for gene transfer, given their higher efficiency. Another approach relies on molecular conjugates, consisting of a polylysine-DNA complex and a macromolecule receptor ligand that can be internalized by the target cells [45]. Lipid nanoparticles (LNPs) are formulated by combining lipid-based components and siRNA or modRNA; the combination of the organic and aqueous components induces hydrophobic/hydrophilic interactions, resulting in the formation of nanoparticles [46]. The lipidoid components can be optimized through combinatorial chemistry approaches to achieve higher stability of the LNPs at 4 °C after synthesis, without requiring freezing temperatures for their transport and storage, thus facilitating their distribution to clinical settings [47]. A study assessing the efficacy of formulated LNPs showed that LNPs remained stable for 15 days in storage at 4 °C after synthesis; although there was a reduction in activity within the first 24 h, the ability to transfect cells in vitro successfully was retained and remained unchanged for the remaining days [48]. This study also demonstrated rapid and highly efficient transfection of cells in vivo in rats and pigs, with peak expression within 20 h of delivery. The expression decreased to an almost negligible level by week 2 after administration, which renders this approach applicable for indications where transient expression is preferred or when repeated administration is feasible [48]. Non-viral vectors are less used for lung delivery. This is a path that remains to be further explored. Another advantage of using nanoparticles for the delivery of RNA-based gene therapy includes the enhancement of cellular uptake, endosomal escape, and protection from nuclease degradation; furthermore, targeted delivery can be achieved by enhancing nanoparticle compatibility through surface modifications. A study tested two polymers to deliver miRNA mimics into CF airway epithelial cells, including PEI and chitosan [49]. While PEI-based nanoparticles were more effective in facilitating miRNA uptake into the epithelial cells than chitosan, both nanoparticles appeared to be nontoxic [49]. Another study used an experimental PAH rat model to assess the therapeutic effects of the intravenous delivery of antisense oligonucleotide against miR-145 (anti-miRNA-145) with loaded LNPs injected three times over 5 weeks. The authors found that anti-miRNA-145 was distributed mainly in the lungs, liver, kidney, and spleen, and no major off-target effects were observed in rats [50]. A recent review summarized the delivery of miRNA by employing various types of nanoparticle-based therapeutics in respiratory diseases, including the common nanoparticles mentioned above and some novel nanoparticle delivery systems [51]. Apart from RNA-based therapies, decoy oligodeoxynucleotides (ODNs) were recently found to be another promising gene therapy agent, especially nuclear factor KB (NFKB) decoy ODNs, which have shown great potential in the treatment of respiratory diseases by reducing the NFkB-mediated inflammatory signaling pathway [52]. Various nanoparticles have been tested to deliver NFkB decoy ODNs both in vitro and in vivo with impressive outcomes. In an in vitro model of CF induced by interleukin-1β or Pseudomonas aeruginosa lipopolysaccharide-stimulated bronchial epithelial cell lines, NFkB decoy ODNs coated with polysialic acid-N-trimethyl chitosan or poly(D,L-lactide-co-glycolide), large porous particles efficiently reduced the secretion of multiple pro-inflammatory mediators of CF [53,54]. Moreover, in a PAH rat model, a single intratracheal instillation of polymeric nanoparticle (NP)-mediated NF-kB decoy resulted in the delivery of NPs into lungs and attenuated the setting of PAH by reducing vascular remodeling and inflammation [55]. More details about NFKB decoy ODN-based gene therapies in respiratory diseases were summarized in a recent review [52].
Recent clinical studies have tested the options of non-viral therapy for respiratory diseases. On the basis of a preliminary clinical trial using the lipid GL67A as the vector, a phase 2B clinical trial was performed to evaluate the efficacy in CF patients, and it revealed a modest benefit on lung function after one year [56,57]. Another study tested DNA nanoparticles with PEG-substituted 30-mer lysine polymers in CF patients, reporting successful transduction efficacy without serious adverse effects [58].
This entry is adapted from the peer-reviewed paper 10.3390/cells11060984
This entry is offline, you can click here to edit this entry!