Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pharmacology & Pharmacy
L-asparaginases (EC 3.5.1.1) are a family of enzymes that catalyze the hydrolysis of L-asparagine to L-aspartic acid and ammonia. These proteins with different biochemical, physicochemical and pharmacological properties are found in many organisms, including bacteria, fungi, algae, plants and mammals. To date, asparaginases from E. coli and Dickeya dadantii (formerly known as Erwinia chrysanthemi) are widely used in hematology for the treatment of lymphoblastic leukemias.
- L-asparaginase
- protein engineering
- substrate specificity
1. Introduction
For more than 50 years, drugs with enzymatic activity have been used in clinical medicine. Replacement therapy for pancreatic insufficiency, acceleration of wound healing or thrombolytic management are among the most successful areas of enzymatic drug use. Enzymes that irreversibly destroy certain vital amino acids are being developed as antitumor therapeutics [1]. The first bacterial enzyme introduced into routine clinical practice was L-asparaginase (L-ASNase, L-asparagine amidohydrolase (EC 3.5.1.1)) [2]. Currently, native L-ASNase from Escherichia coli (EcA) or Dickeya dadantii (formerly known as Erwinia chrysanthemi) (ErA), along with the pegylated form of E. coli asparaginase, are successfully used for the treatment of patients with acute lymphoblastic leukemia [3,4,5,6]. Normal and tumor cells require L-asparagine for their metabolic needs. Normal cells can synthesize L-asparagine for their growth with the aid of asparagine synthetase. Neoplastic cells lack the ability to synthesize asparagine due to the absence or shortage of L-asparagine synthetase and are dependent on an exogenous supply of this amino acid from the bloodstream [7]. The anticancer effect of L-ASNase is based on its ability to hydrolyze L-ASN to L-aspartate and ammonia. The exposure of tumor cells, mainly leukemic cells, to L-ASNase leads to disturbance of protein synthesis and cancer cell starvation, resulting in their death [8]. L-ASNases have been identified in mammals, birds, plants, fungi and a wide range of bacteria [9,10]. To date, dozens of microbial sources of L-ASNases have been revealed, although not all of them demonstrated cytotoxicity against leukemic cells or tumor inhibitory effects [1,10].
L-ASNases have for a long time been classified into three families: the plant type, the Rhizobium etili type and the bacteria type. All bacterial L-ASNases can be subdivided into two types depending on their inducibility, cellular localization, affinity to the substrate and quaternary structure [11]. Type I L-ASNases are constitutively expressed enzymes localized in the cytoplasm and have a relatively high Km for L-asparagine. L-ASNases Bacillus subtilis [12], Pyrococcus horikoshii [13] and Acinetobacter soli [14] are the most studied examples of type I enzymes, which show a relatively low affinity to L-asparagine, resulting in nontherapeutic applications. Type II bacterial L-ASNases are periplasmic enzymes with induced expression during anaerobiosis that have a high affinity for L-asparagine and a wide substrate specificity, resulting in potent antitumor activity [15].
The therapeutic use of L-ASNases is limited by a variety of side effects: hepato- and nephrotoxicity, dysfunctions in the central nervous system, pancreatitis, thromboembolism, mucositis, hyperglycemia and dyslipidemia [16,17,18]. Moreover, genotoxic activity was shown for L-ASNase produced by Streptomyces ansochromogenes [19]. Such side effects are considered to be attributed to non-specific effects of these enzymes. In addition to the well-studied antiproliferative effects of L-ASNases, which are believed to be caused by L-ASN deprivation in the tumor cell environment, several alternative mechanisms have also been suggested. Other L-ASNase substrates include L-glutamine, D-asparagine, succinic acid monoamide and asparaginyl-tRNA [20,21]. Thus, antiproliferative or side effects may appear due to their degradation. In 1970, it was shown that L-ASNase from E. coli may release carbohydrates from the α2-HS-glycoprotein fetuin, suggesting that hydrolysis of cell-membrane glycoproteins and inhibition of their synthesis by the enzyme can result in cell lysis [22]. This enzyme could also inhibit glycoprotein biosynthesis and lead to membrane sensitivity due to the specific effect on the concanavalin A receptor in the sensitive and resistant L5178Y murine lymphoma cell line [23]. A very surprising cytotoxic asparagine-independent mechanism was described for a Rhodospirillum rubrum mutant L-ASNase (RrA). RrA demonstrated regulatory capacity and could suppress telomerase activity in several human cancer cell lines, normal activated CD4+ T lymphocytes and xenografts of human solid tumors [24,25,26]. These observations denote the existence of complex mechanisms of action of at least one L-ASNase to a given cell line.
2. Structures of L-ASNases and the Mechanism of Action
The practical application of L-ASNases is currently directly related to their enzymatic properties. The structural, biophysical and biochemical properties of type I and II L-ASNases have been described in experimental works [11,27,28,29,30], and detailed information about the catalytic mechanism of bacterial L-ASNases is presented in detail in the reviews by Lubkowski et al. [31] and Loch [32].
All known bacterial type II L-ASNases, including those used in medicine, are highly homologous in terms of the amino acid sequence (Figure 1). Such a similarity in the amino acid sequence determines a high degree of similarity in their tertiary and quaternary structures: they fold as homotetramers with four active sites between the N- and C-terminal domains of two adjacent monomers [33,34,35,36,37]. Moreover, bacterial L-ASNases have a high degree of similarity in their tertiary and quaternary structures.

Figure 1. Multiple alignments and conservative residues of the most extensively studied L-ASNases. Multiple sequence-alignment toolbox PRALINE (http://ibivu.cs.vu.nl (accessed 27 May 2021) [38]) was used. Conservative residues were highlighted according to the legend. An asterisk denotes absolutely conserved residues. EcA, Escherichia coli II L-ASNase; ErA, Dickeya dadantii (Erwinia chrysanthemi) L-ASNase; EwA, Erwinia carotovora L-ASNase; HpA, Helicobacter pylori II L-ASNase; RrA, Rhodospirillum rubrum L-ASNase; WsA, Wolinella succinogenes L-ASNase.
Each monomer has 40 β-layers and 8 α-helices arranged in an N-terminal domain and a smaller C-terminal domain connected by a linker of approximately 20 amino acid residues [20,39,40]. The apex of the substrate-binding site is covered by a flexible loop (amino acid residues 14–33), which acts as a “lid”, modulating the affinity to the substrate [41,42]. One of the residues of the lid loop (in the case of EcA–N24) participates in a network of hydrogen bonds near the catalytic site (the critical residue of Y25). In all L-ASNases, the conformational change of the flexible loop after binding of the substrate creates a medium that stabilizes the negatively charged tetrahedral intermediate, thereby ensuring the effectiveness of the enzymatic reaction. From the very beginning, it was clear that catalysis occurs through nucleophilic substitution. However, the question remained: is the reaction a single substitution involving only the activated water molecule, or does it involve two stages of double substitution (also called a ping pong reaction, Figure 2) [43,44]. The mechanism of double substitution with an intermediate acylated enzyme has been challenged by experimental data on mutants T19A, T116A, T19A/T116A, K188M and Y308F of the active site of guinea pig L-ASNase in support of the mechanism of direct displacement of type I/II L-ASNases [34,45]. However, several recent papers have confirmed the original interpretation of the catalytic mechanism [20,31,46], and apparently, the exact mechanisms of enzymatic hydrolysis of asparagine of different classes differ.
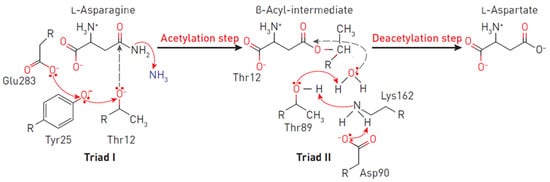
Figure 2. Reaction mechanism at the EcA II catalytic triads. The triad I acylates the substrate (L-asparagine) to form a β-aspartyl enzymatic intermediate. The triad II deacylates the intermediate in the presence of a water molecule to release L-aspartic acid and ammonia as products. In the first reaction, the electron density migrates from Glu283 to the oxygen of the Tyr25, and consequently, to the oxygen Thr12. A nucleophilic attack occurs, leading to the release of ammonia and the formation of ether. In the second reaction, due to the presence of a charge on Asp90, an ionic bond is formed with the amino group Lys162, leading to the removal of the proton from Thr89, followed by the nucleophilic attack of the water molecule on the carbon of the ester. Thus, a deacetylation reaction occurs.
The primary nucleophile of the T12 enzyme was identified after the publication of the EcA structure [36,47]. After the addition of asparagine, the nucleophilic threonine, located close to the active center of the protein, interacts with the carbonyl group of the amide substrate (L-Asn) to form an intermediate product of the reaction—an acyl-enzyme. An ammonia molecule is split from the substrate to form another product of the reaction. The acyl-enzyme reacts with a water molecule to release L-ASNase and form aspartate [40,44,48,49]. Threonine activation is a combination of several coordinated effects. In the EcA enzyme, the primary role is assigned to invariant N90 as it forms a set of hydrogen bonds, which involve Y25 (Figure 3). Additional factors include the precise positioning of the substrate molecule, the participation of the water molecule, and the hydrogen bond that is formed between the nitrogen of the carboxamide group of the substrate and the carbonyl oxygen of the main chain of the enzyme in A114 [8]. The role of the catalytic activity of residues in substrate binding and specificity (T12, Y25, T89, D90 and K162 in EcA) has been established in several studies [8,50,51,52]. These residues are also conserved among other bacterial enzymes (shown in Figure 1). The coordinating role of several motifs in the first nucleophilic attack explains why “catalytically deficient” variants (i.e., lacking either T89 or Y25) may still retain residual catalytic activity. By chemical modification, 1H-nuclear magnetic resonance and 1H-NMR spectroscopy, it was determined that tyrosine and histidine are components of the active center of EcA [53].
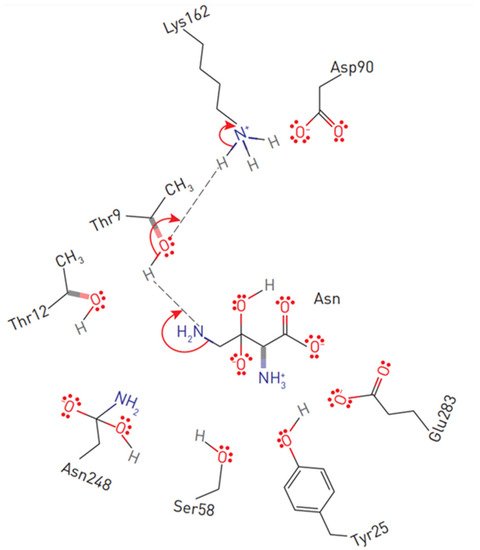
Figure 3. Substrate positioning in EcA II active center. Displayed are conserved active site residues with their relative locations to L-asparagine.
The catalytic mechanism of L-ASNases depends on two highly conserved catalytic triads. The first consists of amino acids threonine, lysine and aspartic acid (in EcA: T12, K162 and D90), resembling serine, histidine and the aspartate triad of classical serine proteases, which mainly participate in the catalysis of the reaction. The second, consisting of threonine, tyrosine and glutamic acid (in EcA: T12, Y25 and E283), participates in the binding of the substrate and the release of reaction products [52,54,55,56]. However, another study demonstrated that E283 is not essential for catalysis [57].
In contrast to the triad functioning in EcA, in the ErA structure, Y29 is well ordered and forms hydrogen bound to T15, but its side chain does not interact with any residue from the second protomer in the dimer (the nearest distance to carboxylate of E289 exceeds 12 Å), which goes against the universal presence of the second catalytic triad (T, Y, E) in L-ASNases [37]. Structural analysis of ErA showed that residues T15, T95, S62, E63, D96 and A20 are in contact with the ligand [20]. The catalytic triads in L-ASNase Helicobacter pylori (HpA) are represented by T16, Y29, E289 and T95-D96, K168 [49]. Replacement of amino acids forming the active center of HpA T16E, T95D and T95H led to a complete loss of enzymatic activity [58]. T95 is not directly involved in catalysis, but it is involved in the activation of the water molecule responsible for the second nucleophilic attack during the deacylation step. T95 is also involved in substrate binding. Replacement of T95 with histidine or aspartate completely disrupts enzymatic catalysis.
Molecular and genetic analysis of E. coli genes encoding L-ASNases I and II, including 5′- and 3′-untranslated regions, revealed one region of sequence similarity and differences in 11 positions [33,59]. A typical secretory signaling peptide of 22 residues was also found, the promoter region was identified and the site of the beginning of transcription of the gene encoding L-ASNase II was determined. Its strict regulation by cyclic AMP receptor protein and anaerobiosis protein, which regulates fumarate and nitrate reductase, was confirmed.
The crystal structure of a human L-asparaginase-like protein was obtained, and by rational engineering, three double mutants and two quadruple mutants were created, which allowed the catalytic activity to be increased up to six times [60]. Structurally, the hASNase3 protein belongs to the N-terminal nucleophile (Ntn) family, which requires autocleavage between G167 and T168 to become catalytically active. To determine the individual contribution of each of the three conserved threonines of the active center (threonine triad T168, T186, T219), mutants T168S, T186V and T219A/V were prepared for enzyme-activating autocleavage reactions, and their ability to cleave and catalyze asparagine hydrolysis was tested. These studies have shown that although not all threonines of the triad are necessary for the cleavage reaction, they are all obligatory for ASNase activity.
The crystalline bacterial enzyme EcAI is organized into a tetrameric assembly, but it is not clear whether the protein exists as a tetramer in solution. The same doubt was expressed about the type I enzyme of archaea from Pyrococcus horikoshi, which acts as a dimer rather than a tetramer [33]. L-ASNases of some extremophile bacterial organisms [61], for example, Thermus thermophilus [62], Thermococus sibiricus [63] or Melioribacter roseus [64], can act as hexamers (trimers of dimers), but there is no structural evidence to support this hypothesis. It is believed that the dimer of L-ASNase II, which has two active centers, is not able to cleave L-asparagine. The effectiveness of the octamer or dodecamer is also reduced, although these functional states of the enzyme can be detected in smaller percentages in commercial preparations, which indicates an issue with the stability of the enzyme.
3. Stability and Activity of L-ASNases
It is generally believed that the stability and activity of enzymes are antagonistic. Several examples support this hypothesis, showing that more stable enzymes tend to exhibit a lower rate of catalyst [65,66]. Substitution E289A in HpA sharply reduced the catalytic activity of the enzyme but increased its thermal stability, which suggests a stabilizing role of such mutation [58]. In the HpA quaternary structure, E289 is located near the active site, close to Q63 from a neighboring monomer and N255 from the same monomer. Amino acids that determine the properties of the most studied L-ASNases and that are subject to molecular substitution are shown in Figure 4. The replacement of E289A in HpA led to a loss of interactions in the interunit space and with the substrate. Moreover, alanine at position 289 interrupted all interactions with N255 and Q63, which may lead to a change in the localization of the variable loop to a conformation more similar to another enzyme from Dickeya dadantii [20,49]. The resulting conformation could be the reason for the mutant’s inability to carry out proper catalysis.
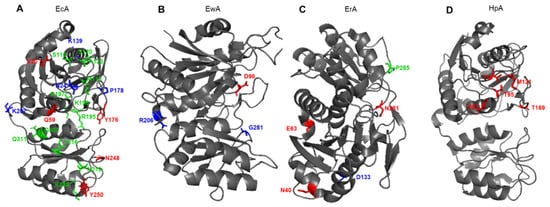
Figure 4. Schematic presentation of amino acid residues that determine the properties of (A) EcA, Escherichia coli L-ASNase II; (B) EwA, Erwinia carotovora (Pectobacterium carotovorum) L-ASNase; (C) ErA, Dickeya dadantii (Erwinia chrysanthemi) L-ASNase; (D) ErA, Helicobacter pylori L-ASNase. Red font shows amino acids associated with L-glutaminase activity. Blue font shows amino acids associated with the stability of the enzyme. Green font shows amino acids associated with immunogenicity.
There are several counterexamples to the rule of antagonistic relations between stability and enzymatic activity [67], and L-ASNase may be one of these exceptions. It was shown that mutations leading to an increase in the flexibility of the loop closing the substrate-binding site reduce the catalytic reaction, whereas mutations stabilizing the loop lead to an increase in the catalytic effect [68]. Variants with increased thermal stability have often improved catalytic efficiency, even if modifications are applied to subunit interfaces and their flanking residues [69,70]. Since part of the binding site is located at the monomer-monomer interface, a possible explanation is that mutations that enhance the interaction of subunits contribute to the tight binding of the substrate and improve the compactness of the tetramer. The conservative mutation A176V in the intracellular L-ASNase of Saccharomyces cerevisiae completely suppressed the activity of the enzyme. Sequences and structural comparisons with bacterial L-ASNases of type II have shown that the mutated residue is located in a highly conserved region and participates in the coupling of functional tetramers [71]. Verma et al. [69], using the example of several variants of EcA with amino acid substitutions, also showed that even small changes at the interface of subunits can significantly affect the stability of EcA. In a study by Vidya et al. [72], it was shown that in EcA, the most stabilizing mutations on the surface loops K139D and K207D caused neutralization and/or inversion of total protein charge and eliminated unfavorable electrostatic interactions compared to the control mutant K139R and the wild-type enzyme. In another study, the Erwinia carotovora variant with a single point mutation D133L had a half-inactivation temperature of 55.8 °C, whereas the wild-type enzyme had a half-inactivation temperature of 46.4 °C [73]. At 50 °C, the half-life values for wild-type enzymes and mutant enzymes were 3 and 160 h, respectively. Screening of the library of random substitutions of the D133 residue and analysis of the electrostatic potential of the wild-type enzyme showed that D133 is located in a neutral region on the surface of the enzyme and makes a significant and unfavorable electrostatic contribution to overall stability. The single-point mutation of N24S led to the creation of an EcA variant with higher thermal stability, which also retained levels of catalytic activity equivalent to the native variant and had less degradation during long-term storage [74]. The binding of L-asparagine to the allosteric site in the crystal structure of EcA I was observed simultaneously with the reorganization of the quaternary structure in this study. The carboxyl group of bound asparagine formed salt bridges and hydrogen bonds with R240, while nitrogen N (delta 2) interacted with T162. The R240A mutation increased the Km from 0.5 to 5.9 mM, presumably due to a decrease in the site’s affinity with L-asparagine. Signal transmission from the allosteric site to the active site appears to involve tiny interactions at the dimer-dimer interface and Q118 position near the active site for the binding of a water molecule.
The importance of the five tyrosine residues of EcA II for its stability and catalysis has been studied extensively by Derst et al. [52] using site-directed mutagenesis, chemical modification and thermodynamic studies of protein denaturation. While tyrosine residue Y25 is directly involved in catalysis, the hydroxyl groups of residues Y181, Y250, Y289 and Y326 were not necessary for the activity of EcA, but residues Y181 and Y326 were involved in the stabilization of the natural tetramer EcA. The pH titration curves showed that the residue of the active center Y25 has a normal pKa, while the C-end of Y326 is unusually acidic. The 1H-NMR signals of a specific ligand-sensitive tyrosine residue were assigned to Y25. None of the mutations H87A, H87L, H87K, H183L or H197L in EcA significantly affected Km in reaction with beta-hydroxamate or binding of aspartate.
The data from Wehner et al. [75] question whether histidine residues are necessary for the catalysis of EcA and suggest that H183 is important for the stabilization of the native ASNase tetramer. 1H-NMR and fluorescence spectroscopy show that H87 is located inside the protein, possibly near the active center. Single-point amino acid substitution of G281S in L-ASNase from Erwinia carotovora significantly affects its structural and functional properties and leads to a 10.8-fold increase in Km and a 45.5-fold decrease in catalytic activity to L-asparagine [76]. The mutant exhibited altered kinetic properties depending on pH and demonstrated an increased temperature of half-inactivation compared to the wild-type protein, which suggests that G281 contributes to the low stability of the enzyme.
Bioinformatical and structural analysis by Li et al. [77] identified structures that affect the thermal stability of thermophilic and nonthermophilic type II L-ASNases. Modification of these structures in L-ASNases from Pyrococcus yayanosii (PyA), Thermococcus gammatolerans, Bacillus subtilis and E. coli made it possible to verify that amino acids at positions 51 and 298 of PyA and correspondingly those at 57 and 305 of EcA are key amino acids responsible for the thermal stability of thermophilic and nonthermophilic type II L-ASNases. Moreover, the tightness of the C-end, the rigidity of the loop and the low surface charge near the active site are of great importance for the thermal stability of L-ASNases.
Using several modeling approaches in silico, Mahboobi et al. [78] developed a new structure of EcA to improve its pharmokinetic profile and predicted an enzyme variant with four mutations, L23G, K129L, S263C, and R291F, with lower toxicity, higher stability, and an increased half-life. L-ASNase from Bacillus licheniformis, when replaced by D103V, demonstrates increased thermal stability, specific activity, affinity to the substrate and a three-fold increased half-life compared to the native enzyme [79]. L-ASNase Pyrococcus furiosus, in the case of K274E mutation, had improved enzymatic properties under physiological conditions and showed approximately 2.5 times higher catalytic efficiency, reduced activation energy and two times less Km at 37 °C compared to the wild-type enzyme [80]. In addition, Gervais and Foote [81] found that two L-ASNase mutants from Dickeya dadantii have a single deamidation site, N41D or N281D, and approximately the same specific activity as the wild type. However, the double-mutant N41D N281D has increased specific activity. Structural analysis showed that small changes caused by the N41D point mutation may have reduced the number of hydrogen bonds in this α-helical part of the protein structure. The N281D mutation resulted in a decrease in glutaminase activity compared to the wild type and the N41D mutant. However, the N281D mutation also gave the enzyme less stability at elevated temperatures. In general, these data suggest that deamidation at sites N41 and N281 does not affect enzyme activity and should not cause concern during processing, storage or clinical use.
According to sequence alignment and homologous modeling, the residues of G107D, T109Q, T109S and S166A of type II L-ASNase from Bacillus subtilis adjacent to the catalytic cavity were replaced by site-directed mutagenesis [70]. The mutant G107D showed increased heat resistance and higher activity with L-asparagine than the wild-type protein. A comparative analysis of the interactions of hydrogen bonds, the surface electrostatic potential and the structure of the substrate-binding pocket between G107D and the wild-type protein showed that G107D replacement led to small conformational changes and redistribution of the surface electrostatic potential, while contributing to improved protein stability and catalytic efficiency.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics14030599
This entry is offline, you can click here to edit this entry!