Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
Rheumatoid arthritis (RA) is a complex and heterogenous disease; what differentiates patients with RA are the number of affected joints, antibodies and serum cytokines levels, and the severity of joint destruction. TNF (tumor necrosis factor) is a proinflammatory cytokine which exerts multiple biological activities in RA. Blocking TNF with antibodies became a standard of modern RA therapy in patients not responding to conventional synthetic disease-modifying antirheumatic drugs (csDMARDs), e.g., methotrexate.
- rheumatoid arthritis
- anti-TNF treatment
1. Introduction
Rheumatoid arthritis (RA) is a complex and heterogenous disease; what differentiates patients with RA are the number of affected joints, antibodies and serum cytokines levels, and the severity of joint destruction [1]. TNF (tumor necrosis factor) is a proinflammatory cytokine which exerts multiple biological activities in RA. Blocking TNF with antibodies became a standard of modern RA therapy in patients not responding to conventional synthetic disease-modifying antirheumatic drugs (csDMARDs), e.g., methotrexate. TNF inhibitors include infliximab, adalimumab, etanercept, golimumab, and certolizumab pegol, each having a similar efficacy in RA. TNF inhibitors remain the most practically useful biological drugs in RA, but their effectiveness is diverse in individual cases, which reflects the heterogeneity of RA.
2. The Pivotal Importance of TNF in RA
TNF is a pleiotropic proinflammatory cytokine which is produced mainly by activated monocytes and macrophages and, to a lesser extent, by T-lymphocytes. The T cell-derived response is thought to be of particular importance in triggering TNF efflux by synovial macrophages [2]. Citrullination of proteins at sites of inflammation is a key process leading to the activation of macrophages in RA. Citrulline-specific T cells display predominately Th1 and Th17 phenotypes, and their effector cytokines are interferon gamma (IFN-γ) and interleukin (IL)-17, which activate macrophages. Macrophages can be also activated by direct contact with T cells [3]. Anti-citrullinated protein antibodies (ACPAs) are implicated in immune complex formation. Sokolove et al. demonstrated that ACPA-IC is an inducer of TNF production by triggering the innate immune receptor Toll-like receptor (TLR) 4- and the crystallizable fragment gamma receptor (FcγR)-dependent signaling in macrophages [4]. In addition, ACPAs selectively activate extracellular signal-regulated kinase 1/2 (ERK1/2) and c-jun N-terminal kinase (JNK), facilitating nuclear factor κB (NFκB) pathways and TNF production [5]. ACPAs can also suppress the expression of microRNA let 7a, which also contribute to enhancing TNF expression [6]. TNF exists in transmembrane and soluble forms which have different biological functions. The soluble TNF (sTNF) is cleaved from transmembrane TNF (tmTNF) by metalloprotease TNF-alpha-converting enzyme (TACE). The effects of TNF are mediated through two distinct receptors: TNF receptor 1 (TNFR1), expressed ubiquitously, and TNFR2, expressed primarily in immune cells, neurons and endothelial cells. TNFR1 is activated by both sTNF and tmTNF, whereas TNFR2 has a high affinity to only membrane-bound forms [7]. Stimulation of TNFR1/2 exerts distinct cellular responses. TNF signaling through both TNFR 1 and TNFR2 leads to the expression of NF-κB and activator protein 1 (AP-1) target genes associated with cell survival. Activation of TNFR1 is thought to primarily induce proinflammatory responses, whereas TNFR2 mostly mediates local homeostatic signals. TNFR1 is also capable of inducing cell death responses, including apoptosis (programmed cell death) and necroptosis (uncontrolled cell death). Apoptosis, mediated by the caspase-8 activation pathway, is thought to induce paradoxical anti-inflammatory and immunosuppressive responses. In contrast, necroptosis triggers local inflammation [8]. The factors determining whether the pathway leading to cell survival, apoptosis or necrosis is activated, is not well understood. Nevertheless, the ubiquitination status of receptor-interacting serine/threonine protein kinase 1 (RIPK1) seems to be crucial. The ubiquitylated RIPK1 leads to pro-survival signaling, whereas non-ubiquitylated RIPK1 induces either apoptosis or necroptosis. The long form of the FLICE-inhibitory protein, also known as c-FLIP(L), is another important regulator of both apoptosis and necrosis pathways. c-FLIP(L) is a major apoptosis inhibitor, similar in structure to caspase-8, which is also capable of forming a proteolytically active complex with caspase-8, which cleaves RIPK1 and RIPK3, and blocks RIPK1/RIPK3/MLKL-dependent necroptosis [9].
3. Diverse Influence of TNF on Target Cells in RA
3.1. Fibroblast-like Synoviocytes
TNF is an extraordinarily pleiotropic cytokine influencing different types of cells (Figure 1). Synovium is the principal site of immune disorders and inflammation in RA, and consists of the continuous surface layer of cells (intima) and the underlying tissue (subintima). The healthy synovial membrane contains relatively few cells and is predominantly composed of two types of synoviocytes, forming one to two cells per layer: type A are macrophage-like synoviocytes (MLS), originating in the bone marrow, capable of phagocytosis, whereas type B, also known as fibroblast-like synoviocytes (FLS), are responsible for synthesis of the synovial fluid hyaluronan [10]. In healthy synovium, type A and type B synoviocytes exist in relatively equal proportions. The extension of synoviocytes in RA form the locally invasive synovial tissue, called the pannus [11]. The synovial lining is greatly hypertrophied, and both types of synoviocytes proliferate, but MLS are more abundant [12]. The role of MLS in RA is not entirely clear, but presumably MLS induce invasiveness of FLS via the secretion of proinflammatory cytokines, including TNF, which mostly contribute to joint destruction.
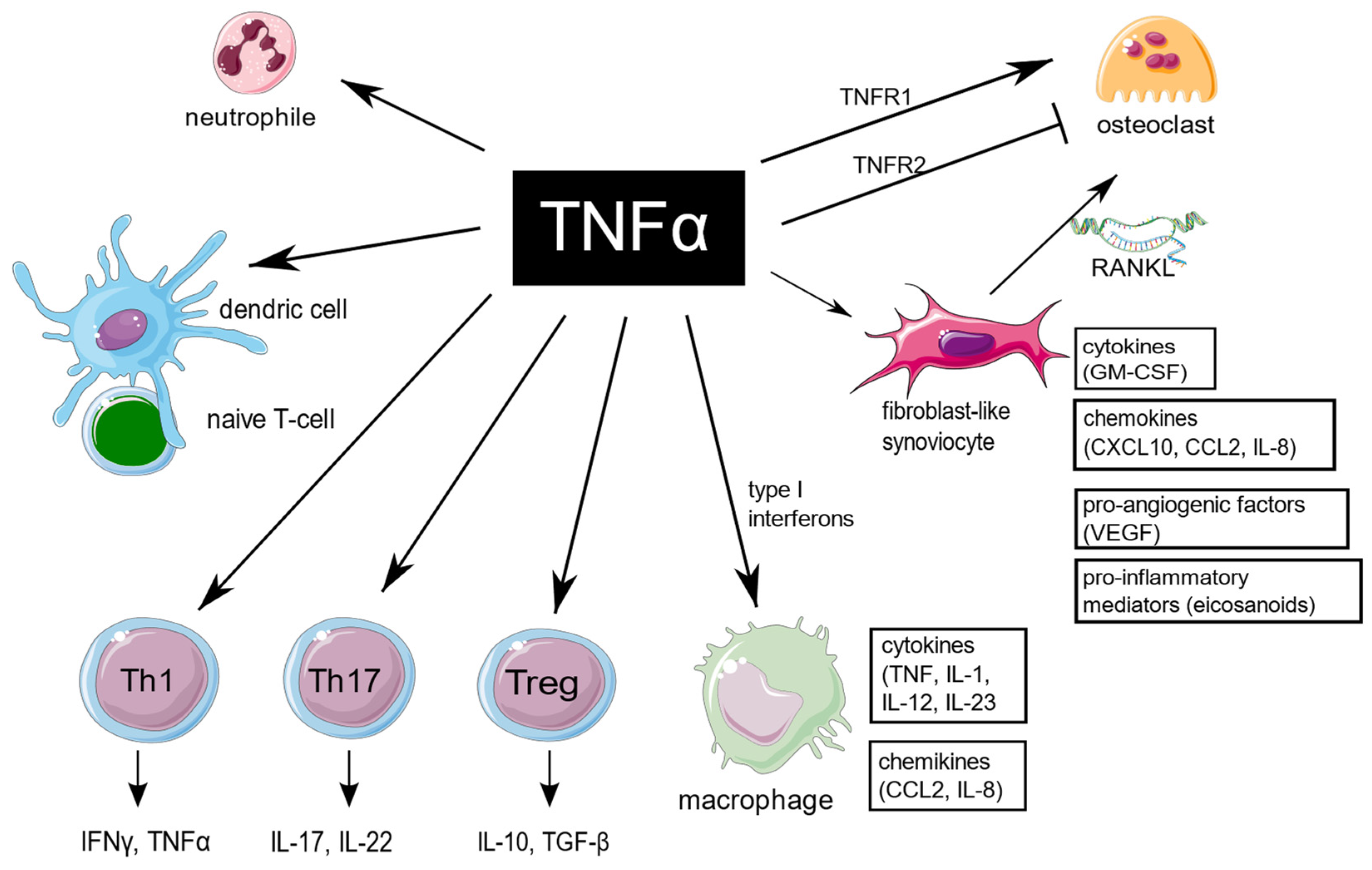
Figure 1. TNF is a pleiotropic cytokine, which is implicated in function of different cells which are important for RA pathogenesis. TNF promote expression of proinflammatory genes in fibroblast-like synoviocytes and macrophages. It has a large impact on T cells by expansion of Th17 lymphocytes subset, transition of Th17 to the non-classic Th1 subset in inflammatory sites, as well as decreasing Tregs levels, and it also enhances leukocyte influx to sites of inflammation. In addition, TNF stimulates neutrophils to delay apoptosis, induce respiratory burst, and upregulate cytokine production. TNF is of key importance in osteoclastogenesis and RA-associated bone loss. The figure was created using the Servier Medical Art template; https://smart.servier.com, accessed on 21 January 2022.
TNF-mediated signaling in FLS is presumably crucial for arthritis initiation [13][14]. FLS are a major source of multiple proinflammatory cytokines and chemokines, which lead to recruitment of more immune cells. At the transcriptional level, TNF in RA regulates the expression of proinflammatory genes in a cell type-specific manner. The TNF-inducible proinflammatory genes exhibit unremitting expression in FLS. The sustained expression in FLS is presumably a result of an alteration of the chromatin structure in regulatory elements of the genes, resulting in the continuous upstream activation of NF-κB and MAPK signaling.
3.2. Macrophages
In macrophages, in contrast to FLS, the proinflammatory genes are only transiently expressed in response to TNF [15]. Moreover, on a prolonged exposure to TNF, macrophages display tolerance induction [16]. However, such desensitization may be blocked by type I interferons. TNF and type I IFNs presumably cooperatively regulate chromatin accessibility at inflammatory gene promoters, facilitating robust transcriptional responses to weak signals [17]. In addition, in macrophages and endothelial cells, TNF induce the IFN-β autocrine loop via activation of interferon regulatory factor 1 (IRF1) and interferon regulatory factor 3 (IRF3) [18]. Treatment with anti-TNF antibodies strongly modulate functions of macrophages in inflammatory conditions. The process is IL-10/STAT3 pathway–dependent. Upon anti-TNF blocking, the surface expression of pro-inflammatory markers (CD40 and CD60) is downregulated, whereas expression of phagocytic receptors (CD16, CD163, MER proto-oncogene tyrosine kinase) is increased. In addition, anti-TNF agents also suppress production of pro-inflammatory cytokines (IL-6, IL-12, TNF) in macrophages [15].
3.3. Bone Marrow Stromal Cells and Osteoclastogenesis
RA-associated bone loss is a result of the disturbance between bone formation and bone resorption. Proinflammatory cytokines, including TNF, are essential for osteoclastogenesis. TNF stimulate the expression of receptor activator of NF-κB ligand (RANKL) by bone marrow stromal cells, mainly fibroblasts and activated synovial T cells. RANKL is crucial for propagating osteoclasts maturation. TNF can also stimulate osteoclast precursors directly through TNFR1 signalling. The soluble TNF is responsible for mobilizing osteoclasts from the bone marrow [19][20]. In addition, TNF is a potent inhibitor of osteoblast differentiation [21][22]. In contrast, TNFR2-dependent signalling inhibits osteoclastogenesis [23].
3.4. Neutrophils
In RA, TNF exposure on neutrophils up-regulates several genes involved in the NF-κB signaling pathway, particularly TNF itself. In addition, TNF induces expression of anti-apoptotic genes, e.g., TNFAIP3 (A20), BCL2A1, CFLAR (FLIP), and FAS, while expression of some pro-apoptotic genes, including APAF1, CASP8, CASP10, FADD, TNFRSF1A and TNFRSF1B, is down-regulated. These transcriptional changes allow neutrophils to delay apoptosis and thereby enhance inflammation. Interestingly, these effects of TNF on human neutrophils seem to be concentration-dependent, since high concentrations may paradoxically promote apoptosis. During continuous exposure to TNF, desensitization of TNF-inducible expression in neutrophiles occurs, which is presumably a result of a down-modulation of both TNFR1 and TNFR2 receptors [24].
3.5. T Cells
The relationship between TNF and T cell responses is complex, since T cells both respond to and produce TNF. TNF is of a key importance for proper T cells development. It is responsible for modulation of T cells development in thymus, as well as generation of efficient adaptive immune responses in lymph nodes. TNF is necessary for the activation of effector, memory, and regulatory T cells. Extensive production of TNF influence maturation of the antigen-presenting dendritic cells, and thus recalls efficient activation of naïve T cells. In addition, TNF was experimentally shown to promote leukocyte adhesion and diapedesis, which enable T cells invading the inflamed tissue [25].
Expansion of Th17 cell subset is hypothesized to be an important mechanism of peripheral tolerance breakdown in RA. The increase in Th17 was reported in RA peripheral blood, synovial fluid and tissue, however Th17 lymphocytes are rarely found at inflammatory sites in comparison with other T cell subsets. This is presumably due to the rapid transition of Th17 lymphocytes to the non-classic Th1 phenotype, induced by TNF. Shifted Th17 cells become particularly aggressive and presumably are of particular importance in pathogenesis of inflammatory disease, including RA [26].
Much knowledge about the influence of TNF on lymphocytes in RA is provided also by data from studies evaluating the effect of treatment with anti-TNF drugs. Interestingly, in long-term observation of anti-TNF-treated patients, the number of circulating Th1, Th2 and Th17 lymphocytes was reported to increase [27][28]. Non-responders are also associated also with higher baseline Th17 frequencies, what may be a result of the impaired peripheral tolerance and paradoxical activation of Th1 and Th17 responses in this group of RA patients [29][30].
Dulic et al. also showed that, as compared with non-responders, anti-TNF responders in long-term observation were characterized with lower proportions of total CD4+ T cells and higher proportion of late activation CD4+HLA-DR+ T cells. In the same study, T cells expressing the early activation marker CD69 were indicated as promising predictive marker of anti-TNF blocker response [27].
TNF appears to inhibit the suppressive functions of Tregs, which are essential in preventing autoimmunity. Tregs prevent optimal costimulation by antigen presenting cells, and further response related to cytotoxic T cells [31]. The mechanism of such TNF-mediated inhibition is downmodulating the expression of the key transcription factor FoxP3. In line with the above, the anti-TNF treatment has a potential to restore the suppressive function in Tregs. The ratio of Th17/Treg in anti-TNF-treated patients, regardless of the effectiveness of therapy, tends to approach the ratio observed in healthy controls [27][32]. Under certain conditions, TNF may also increase Tregs differentiation and promote immunosuppressive effects, which appears to be TNFR2-dependent. The expression of TNFR2 on Tregs is decreased in autoimmune diseases, limiting the importance of this effect in RA. Based on these findings, the development of new anti-TNF therapies should be focused on a more selective blockade of TNFR1-dependent signaling [31].
The TNF inhibitor golimumab was shown to largely affect peripheral memory T cell responses. In long-term observation, patients treated with golimumab displayed increased effector memory T cells and antigen-specific T cell cytokine production [33].
As for B cells, conflicting results have been reported. TNF blocking with infliximab was found to increase the frequencies of circulating memory B-cells, whereas another study with etanercept showed the opposite effect [34].
3.6. Effects of TNF on Angiogenesis
Formation of new capillaries from existing vessels, termed angiogenesis, is a key event in the formation and maintenance of pannus [35]. Proinflammatory cytokines, including TNF, stimulate synovial fibroblasts to release vascular endothelial growth factor (VEGF). VEGF stimulates endothelial cell proliferation, and thus angiogenesis. TNF also affects the later stage of angiogenesis by the increased production of angiopoietin 1, important for newly-formed blood vessel stability [36].
3.7. Influence of TNF on Endothelial Dysfunction
TNF contributes to the increased risk of cardiovascular comorbidities in RA patients. Vascular injury is a result of the disruption of the endothelial barrier. TNF via TNFR1 signaling destabilizes the endothelial skeleton by actin rearrangement and microtubule destabilization [37]. In addition, TNF was shown to induce degradation of the endothelial glycocalyx [38]. TNF also impairs nitric oxide formation, which is of key significance for endothelium-dependent vasodilatation. TNF-signalling decreases NO generation by TNFR1-dependent inhibition of endothelial NO synthase (eNOS) expression and accumulation of the endogenous eNOS inhibitor, but also influences the enhanced removal of NO. Moreover, TNF stimulates production of vascular reactive oxygen species (ROS) by upregulated expression of NADPH-dependent oxidase (NOX) subunits. Upon TNF activation, ROS contribute to TNF-induced NF-kB activation, decreased NO bioavailability and progression of atherosclerosis [39].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23042366
References
- Scott, D.L.; Wolfe, F.; Huizinga, T.W. Rheumatoid arthritis. Lancet 2010, 376, 1094–1108.
- Tran, C.N.; Lundy, S.K.; Fox, D.A. Synovial biology and T cells in rheumatoid arthritis. Pathophysiology Off. J. Int. Soc. Pathophysiol. 2005, 12, 183–189.
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17.
- Sokolove, J.; Zhao, X.; Chandra, P.E.; Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcgamma receptor. Arthritis Rheum. 2011, 63, 53–62.
- Lu, M.C.; Lai, N.S.; Yin, W.Y.; Yu, H.C.; Huang, H.B.; Tung, C.H.; Huang, K.Y.; Yu, C.L. Anti-citrullinated protein antibodies activated ERK1/2 and JNK mitogen-activated protein kinases via binding to surface-expressed citrullinated GRP78 on mononuclear cells. J. Clin. Immunol. 2013, 33, 558–566.
- Lai, N.S.; Yu, H.C.; Yu, C.L.; Koo, M.; Huang, H.B.; Lu, M.C. Anti-citrullinated protein antibodies suppress let-7a expression in monocytes from patients with rheumatoid arthritis and facilitate the inflammatory responses in rheumatoid arthritis. Immunobiology 2015, 220, 1351–1358.
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62.
- Gregory, C.D.; Devitt, A. The macrophage and the apoptotic cell: An innate immune interaction viewed simplistically? Immunology 2004, 113, 1–14.
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8, 111.
- Smith, M.D. The normal synovium. Open Rheumatol. J. 2011, 5, 100–106.
- Bromley, M.; Woolley, D.E. Histopathology of the rheumatoid lesion. Identification of cell types at sites of cartilage erosion. Arthritis Rheum. 1984, 27, 857–863.
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255.
- Armaka, M.; Apostolaki, M.; Jacques, P.; Kontoyiannis, D.L.; Elewaut, D.; Kollias, G. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J. Exp. Med. 2008, 205, 331–337.
- Efimov, G.A.; Kruglov, A.A.; Khlopchatnikova, Z.V.; Rozov, F.N.; Mokhonov, V.V.; Rose-John, S.; Scheller, J.; Gordon, S.; Stacey, M.; Drutskaya, M.S.; et al. Cell-type-restricted anti-cytokine therapy: TNF inhibition from one pathogenic source. Proc. Natl. Acad. Sci. USA 2016, 113, 3006–3011.
- Degboe, Y.; Rauwel, B.; Baron, M.; Boyer, J.F.; Ruyssen-Witrand, A.; Constantin, A.; Davignon, J.L. Polarization of Rheumatoid Macrophages by TNF Targeting Through an IL-10/STAT3 Mechanism. Front. Immunol. 2019, 10, 3.
- Loh, C.; Park, S.H.; Lee, A.; Yuan, R.; Ivashkiv, L.B.; Kalliolias, G.D. TNF-induced inflammatory genes escape repression in fibroblast-like synoviocytes: Transcriptomic and epigenomic analysis. Ann. Rheum. Dis. 2019, 78, 1205–1214.
- Park, S.H.; Kang, K.; Giannopoulou, E.; Qiao, Y.; Kang, K.; Kim, G.; Park-Min, K.H.; Ivashkiv, L.B. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat. Immunol. 2017, 18, 1104–1116.
- Muskardin, T.L.W.; Niewold, T.B. Type I interferon in rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 214–228.
- Abu-Amer, Y.; Erdmann, J.; Alexopoulou, L.; Kollias, G.; Ross, F.P.; Teitelbaum, S.L. Tumor necrosis factor receptors types 1 and 2 differentially regulate osteoclastogenesis. J. Biol. Chem. 2000, 275, 27307–27310.
- Li, P.; Schwarz, E.M.; O’Keefe, R.J.; Ma, L.; Boyce, B.F.; Xing, L. RANK signaling is not required for TNFalpha-mediated increase in CD11(hi) osteoclast precursors but is essential for mature osteoclast formation in TNFalpha-mediated inflammatory arthritis. J. Bone Miner. Res. Off. J. Am. Soc. Bone Mineral. Res. 2004, 19, 207–213.
- Gilbert, L.C.; Rubin, J.; Nanes, M.S. The p55 TNF receptor mediates TNF inhibition of osteoblast differentiation independently of apoptosis. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1011–E1018.
- Zhao, B. TNF and Bone Remodeling. Curr. Osteoporos. Rep. 2017, 15, 126–134.
- Zhang, Y.H.; Heulsmann, A.; Tondravi, M.M.; Mukherjee, A.; Abu-Amer, Y. Tumor necrosis factor-alpha (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type 1 receptor and RANK signaling pathways. J. Biol. Chem. 2001, 276, 563–568.
- Chiewchengchol, D.; Wright, H.L.; Thomas, H.B.; Lam, C.W.; Roberts, K.J.; Hirankarn, N.; Beresford, M.W.; Moots, R.J.; Edwards, S.W. Differential changes in gene expression in human neutrophils following TNF-alpha stimulation: Up-regulation of anti-apoptotic proteins and down-regulation of proteins involved in death receptor signaling. Immun. Inflamm. Dis. 2016, 4, 35–44.
- Jaczewska, J.; Abdulreda, M.H.; Yau, C.Y.; Schmitt, M.M.; Schubert, I.; Berggren, P.O.; Weber, C.; Koenen, R.R.; Moy, V.T.; Wojcikiewicz, E.P. TNF-alpha and IFN-gamma promote lymphocyte adhesion to endothelial junctional regions facilitating transendothelial migration. J. Leukoc. Biol. 2014, 95, 265–274.
- Cosmi, L.; Liotta, F.; Maggi, E.; Romagnani, S.; Annunziato, F. Th17 and non-classic Th1 cells in chronic inflammatory disorders: Two sides of the same coin. Int. Arch. Allergy Immunol. 2014, 164, 171–177.
- Dulic, S.; Vasarhelyi, Z.; Sava, F.; Berta, L.; Szalay, B.; Toldi, G.; Kovacs, L.; Balog, A. T cell Subsets in Rheumatoid Arthritis Patients on Long-Term Anti-TNF or IL-6 Receptor Blocker Therapy. Mediat. Inflamm. 2017, 2017, 6894374.
- Aerts, N.E.; Ebo, D.G.; Bridts, C.H.; Stevens, W.J.; De Clerck, L.S. T cell signal transducer and activator of transcription (STAT) 4 and 6 are affected by adalimumab therapy in rheumatoid arthritis. Clin. Exp. Rheumatol. 2010, 28, 208–214.
- Hull, D.N.; Cooksley, H.; Chokshi, S.; Williams, R.O.; Abraham, S.; Taylor, P.C. Increase in circulating Th17 cells during anti-TNF therapy is associated with ultrasonographic improvement of synovitis in rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 303.
- Talotta, R.; Berzi, A.; Atzeni, F.; Batticciotto, A.; Clerici, M.; Sarzi-Puttini, P.; Trabattoni, D. Paradoxical Expansion of Th1 and Th17 Lymphocytes in Rheumatoid Arthritis Following Infliximab Treatment: A Possible Explanation for a Lack of Clinical Response. J. Clin. Immunol. 2015, 35, 550–557.
- Kruglov, A.; Drutskaya, M.; Schlienz, D.; Gorshkova, E.; Kurz, K.; Morawietz, L.; Nedospasov, S. Contrasting contributions of TNF from distinct cellular sources in arthritis. Ann. Rheum. Dis. 2020, 79, 1453–1459.
- Valencia, X.; Stephens, G.; Goldbach-Mansky, R.; Wilson, M.; Shevach, E.M.; Lipsky, P.E. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood 2006, 108, 253–261.
- Khanniche, A.; Zhou, L.; Jiang, B.; Song, J.; Jin, Y.; Yin, J.; Wang, S.; Ji, P.; Shen, H.; Wang, Y.; et al. Restored and Enhanced Memory T Cell Immunity in Rheumatoid Arthritis After TNFalpha Blocker Treatment. Front. Immunol. 2019, 10, 887.
- Pala, O.; Diaz, A.; Blomberg, B.B.; Frasca, D. B Lymphocytes in Rheumatoid Arthritis and the Effects of Anti-TNF-alpha Agents on B Lymphocytes: A Review of the Literature. Clin. Ther. 2018, 40, 1034–1045.
- Clavel, G.; Bessis, N.; Boissier, M.C. Recent data on the role for angiogenesis in rheumatoid arthritis. Joint Bone Spine 2003, 70, 321–326.
- Szekanecz, Z.; Koch, A.E. Targeting Angiogenesis in Rheumatoid Arthritis. Curr. Rheumatol. Rev. 2008, 4, 298–303.
- Petrache, I.; Birukova, A.; Ramirez, S.I.; Garcia, J.G.; Verin, A.D. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am. J. Respir. Cell Mol. Biol. 2003, 28, 574–581.
- Chappell, D.; Hofmann-Kiefer, K.; Jacob, M.; Rehm, M.; Briegel, J.; Welsch, U.; Conzen, P.; Becker, B.F. TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res. Cardiol. 2009, 104, 78–89.
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261.
This entry is offline, you can click here to edit this entry!