Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, Research & Experimental
Amyotrophic lateral sclerosis (ALS) is a rapidly debilitating fatal neurodegenerative disorder, causing muscle atrophy and weakness, which leads to paralysis and eventual death. ALS has a multifaceted nature affected by many pathological mechanisms, including oxidative stress (also via protein aggregation), mitochondrial dysfunction, glutamate-induced excitotoxicity, apoptosis, neuroinflammation, axonal degeneration, skeletal muscle deterioration and viruses. This complexity is a major obstacle in defeating ALS.
- amyotrophic lateral sclerosis (ALS)
- oxidative stress
- protein aggregation
- glutamate excitotoxicity
- apoptosis
- neuroinflammation
- axonal degeneration
1. Introduction
Amyotrophic Lateral Sclerosis (ALS), also known as Lou Gehrig’s or Charcot disease, is characterized by progressive deterioration of the upper and lower motor neurons in the brain and spinal cord, which leads to muscle weakness, paralysis and, finally, death due to respiratory failure within three to five years after the onset of the symptoms. ALS generally starts in limb or bulbar muscles, then spreads to other body parts and culminates in respiratory muscle dysfunction [1][2]. The primary symptoms of ALS due to motor neuronal degeneration are fasciculation, muscle cramps and stiffness, dysarthria, dysphagia, emotional lability (pseudobulbar affect (PBA)), which is characterized by uncontrolled laughter or crying, and dyspnea. Furthermore, sialorrhea usually occurs in ALS patients owing to dysphagia, and increased saliva production can result in aspiration pneumonia. In general, ALS patients also suffer from depression and anxiety [3][4].
2. Therapeutic Strategies for ALS Targets
2.1. Therapeutic Strategies against Oxidative Stress
Oxidative stress is the consequence of the imbalance between the generation of reactive oxygen species (ROS), such as hydrogen peroxide (H2O2), superoxide anions (O2−) and hydroxyl radicals (OH−), and the ability of antioxidant defence system to clean or repair the existing damage to proteins and/or DNA. Normally, SOD1 converts the superoxide anion to H2O2, but as the SOD1 mutation shows lower affinity for Zn2+, it donates an electron to O2 to generate O2− at the Cu2+ catalytic site as a stronger oxidant and reacts with nitric oxide (NO) to form peroxynitrite (ONOO−), which is very detrimental to CNS. ROS is considered to be a major mechanism in ALS causing motor neuron death, whereas some studies imply that ROS only exacerbates disease progression. When glutamate receptors are over-activated in the presence of higher glutamate levels in the synapse, elevated calcium influx into the cell also triggers the entry of calcium into the mitochondria, causing mitochondrial dysfunction, further ROS production and, ultimately, cell death. Glial and infiltrated immune cells also abundantly contribute to the production of ROS because glial synapses, which surround the devastated neurons, stimulate glutamate excitotoxicity and elevated calcium entry into the cell and mitochondria. As a result, each process in neuronal degeneration repeats itself, like a vicious circle [5][6][7][8].
Since lipids constitute polyunsaturated fatty acids, they are prone to oxidize to peroxyl radicals, resulting in cerebral ischemia. As phenol derivatives are well-known radical scavenging agents, Mitsubishi Tanabe Pharma Corporation developed potential phenol-like radical scavengers for the treatment of cerebral infarction. They designed compounds carrying a carbonyl group, which can easily convert to hydroxy group by keto-enol tautomerization to achieve radical scavenging activity, similar to that of the phenol provided by its hydroxy group (Figure 1a). They identified edaravone as an effective radical scavenger among a variety of compounds. Edaravone is capable of scavenging both lipid- and water-soluble peroxyl radicals as well as ONOO− among many types of ROS. Researchers also reported that the in vitro lipid peroxidation inhibitory activity of edaravone was reduced with the substitution of polar or hydrophilic groups in the 2-pyrazoline-5-one ring, while this activity was increased with the lipophilic substitutions on the phenyl ring (Figure 1b) [9][10]. Edaravone interacts with both peroxyl (LOOꞏ) and hydroxyl (ꞏOH) radicals by means of its enolate form (B), followed by the formation of a stable oxidation product (OPB: 2-oxo-3-(phenylhydrazono)-butanoic acid) through a radical intermediate (Figure 2) [11].
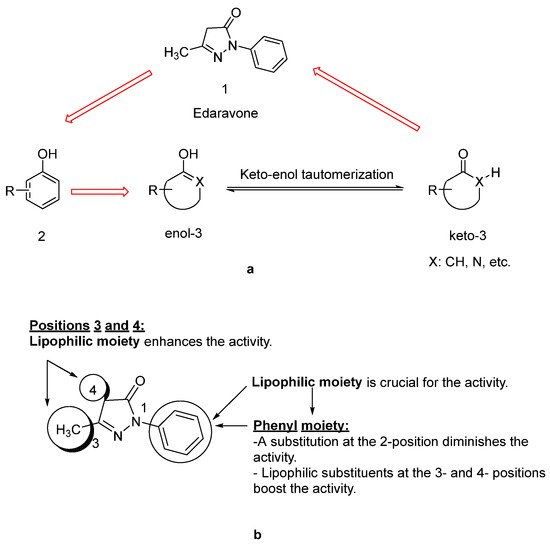
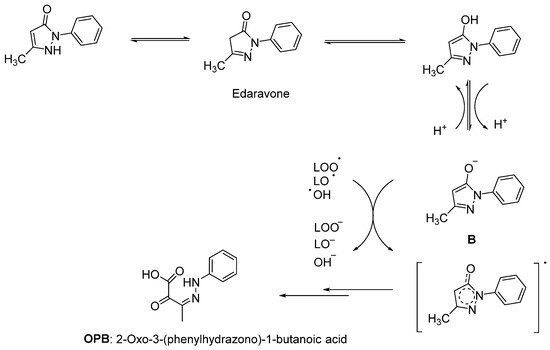
Figure 2. Reaction mechanism of edaravone with free radicals [11].
Edaravone (MCI-186, 3-methyl-1-phenyl-2-pyrazoline-5-one, Radicut®, Radicava®), a neuroprotective antioxidant drug, was synthesized via the reaction of phenylhydrazine and acetoacetic ester by Knorr in 1883 [10]. This procedure was also applied in other studies, as follows: Phenylhydrazine and methyl acetoacetate were mixed in the presence of glacial acetic acid and stirred at reflux for 3 h. The reaction mixture was evaporated to dryness and the residue was extracted with a mixture of water and ethyl acetate. The organic layer was separated, while the aqueous layer was further extracted with ethyl acetate. The combined organic layer was dried over anhydrous sodium sulphate and concentrated over reduced pressure to afford a pure product as a white solid (Figure 3) [12][13]. Edaravone (Figure 4) has been used to treat acute-phase cerebral infarction for almost 20 years in Japan. It received its approval for the treatment of ALS in Japan and South Korea in 2015; the FDA approved the drug in 2017 and Chinese–NMPA in 2019. In a phase II trial, 30 mg or 60 mg of edaravone was administered to 20 subjects with ALS. It was observed that the decline in the amyotrophic lateral sclerosis functional rating scale (ALSFRS-R) score was significantly reduced during the six-month treatment period [14]. In a randomised, double-blind phase III study, a significantly smaller decline in the ALSFRS-R score compared with placebo group was also observed [15]. The exact mechanism of action of edaravone for ALS remains unclear; it has both neuroprotective effects against oxidative stress and anti-inflammatory properties against activated microglial cells. So far, there has been no oral dosage formulation of edaravone clinically, thus it is used as an intravenous therapeutic for ALS management. However, oral dosage of edaravone for ALS patients is performed by Mitsubishi Tanabe Pharma Corporation under a phase 3b, multicentre, randomized, double-blind test, which has enrolled 380 ALS patients to examine and compare the efficacy of two dosing regimens of oral edaravone (Clinicaltrial.gov NCT04569084) [16][17][18].
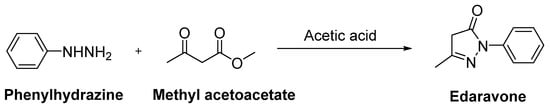
Figure 3. Synthetic route for edaravone.
Figure 4. Therapeutics against oxidative stress.
2.2. Therapeutic Strategies against Oxidative Stress via Protein Aggregation
SOD1 is a Cu/Zn-dependent antioxidant enzyme that catalyses the conversion of superoxide radicals to oxygen and hydrogen peroxide, thus regulating the superoxide levels that arise from mitochondrial inter-membrane space, cytosol and peroxisome. The discovery of the connection between mutations in the SOD1 gene with certain forms of fALS made a tremendous impact in understanding the pathology of ALS. SOD1 mutations have been reported to contribute to ALS through not only protein misfolding and aggregation, but also proteasome impairment, oxidative stress, oligodendrocyte degeneration and mitochondrial dysfunction [19][20].
In order to decrease SOD1 expression and increase SOD1 consumption, antisense oligonucleotides (ASOs) and heat shock protein (HSP) inducers were pursued, respectively. ASOs are short, synthetic nucleic acids which are suitable for chemical modification for high stability in biological fluids and potency in binding their mRNA target. ASOs targeting SOD1 mRNA reduced both SOD1 protein and mRNA levels throughout the brain and spinal cord in the SOD1G93A mice model [21]. Tofersen (BIIB067, ISIS 333611, IONIS-SOD1Rx) is a type of antisense therapy. A randomised, placebo-controlled, first-in-man phase I clinical trial with SOD1-positive ALS patients indicated that ISIS 333611 was well-tolerated when administered as an intrathecal infusion [22]. A second phase I/II trial with BIIB067 was designed to decrease SOD1 mRNA in ALS patients carrying a SOD1 gene mutation. Results showed that a statistically significant reduction in cerebrospinal fluid (CSF)–SOD1 suggested substantial reduction in CNS tissue SOD1. Besides reduced CSF phosphorylated heavy neurofilament, a decline in ALSFRS-R scores were also detected in ALS patients [23]. A phase III trial of BIIB067 is currently ongoing (Clinicaltrial.gov NCT02623699).
HSPs such as Hsp27, Hsp40 and Hsp70 were associated with mutant SOD1 aggregates in SOD1 rodent models of ALS. Arimoclomol (Figure 5), a hydroxylamine-based HSP amplifier, was determined to extend survival and delay disease onset with improvement in neuromuscular activity in the SOD1G93A mouse model of ALS [24][25]. A double-blind, randomized, placebo-controlled phase II trial enrolling patients with rapidly progressive SOD1-mutant ALS suggested a possible therapeutic benefit of arimoclomol [26]. A randomized, double blind, placebo-controlled phase III trial has been completed, but results are not yet posted (Clinicaltrial.gov NCT03491462) [27]. Furthermore, HSPB8, a specific chaperone, was found to be effective in stimulating the clearance of the misfolded proteins related to motor neuron diseases, such as mutant SOD1 and a TDP-43 fragment in ALS. Colchicine (Figure 5), an anti-inflammatory drug generally used in gout treatment, enhances the expression of HSPB8. Colchicine was shown to reduce the aggregation of TDP-43 misfolded species responsible for motor neuron death in sALS [28]. A randomised, double-blind, placebo-controlled, multicentre phase II clinical trial is ongoing for colchicine (Clinicaltrial.gov NCT03693781) [29].
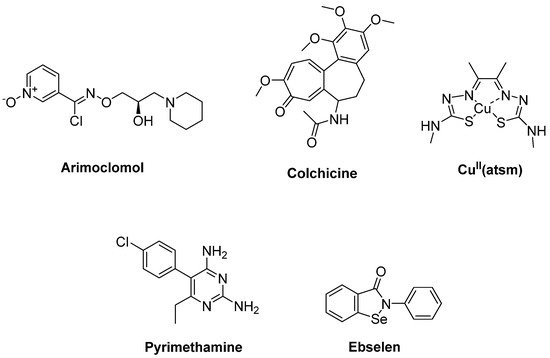
Figure 5. Therapeutics against oxidative stress via protein aggregation.
2.3. Therapeutic Strategies against Mitochondrial Dysfunction
Mitochondria are essential cell organelles that elicit adenosine triphosphate (ATP) through oxidative phosphorylation, regulate calcium homeostasis and produce ROS as a by-product of the electron transport chain. Dysfunction of this electron transport chain can cause increased levels of mitochondrial oxygen consumption and ROS production along with decreased ATP synthesis and DNA repair. Mitochondrial dysfunction also triggers oxidative stress and glutamate excitotoxicity in ALS. Abnormal morphology and ALS-linked mutant proteins were observed in mitochondria of sALS post-mortem tissue [6][7][30].
Olesoxime (TRO19622) (Figure 6) is a mitochondrial pore modulator with a cholesterol-like structure. Bordet et al. [31] determined that olesoxime in vitro promoted motor neuron survival among a collection of a great number of low molecular-weight compounds which were capable of preventing motor neuron cell death. This compound was also found to improve motor performance, delay the onset of the clinical disease and extend survival in SOD1G93A transgenic mice. They also suggested that olesoxime displayed neuroprotective activity, binding to the mitochondrial permeability transition pore (mPTP). However, a phase II–III trial of olesoxime in subjects with ALS revealed that olesoxime did not show a significant beneficial effect in ALS patients treated with riluzole [32].
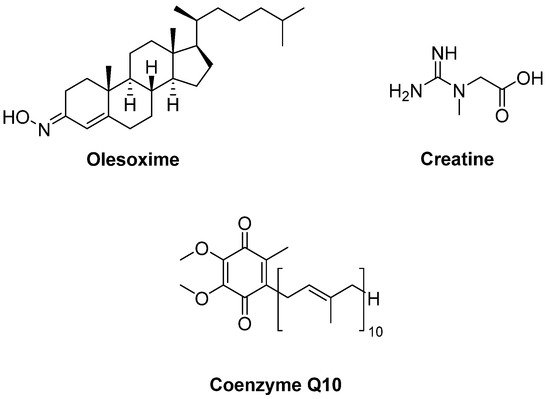
Figure 6. Therapeutics against mitochondrial dysfunction.
Creatine (Figure 6), phosphorylated by creatine kinase, is involved in ATP production. Therefore, creatine stabilizes the mitochondrial creatine kinase and inhibits opening of the mPTP. It was detected that creatine improved motor performance and extended survival in SOD1G93A mice [33]. However, a double-blind, placebo-controlled, sequential clinical trial established that creatine monohydrate showed no beneficial effect on survival or disease progression in ALS patients [34]. Creatine also displayed no benefit in any outcome measure in a randomized double-blind, placebo-controlled trial on 104 ALS patients [35] or in another multicentre, double-blinded study with 107 ALS patients [36]. Coenzyme Q10 (CoQ10) (Figure 6), an electron acceptor in the mitochondrial respiratory chain, was reported to increase lifespan in SOD1G93A mice [37]. In a two-stage, bias-adjusted, randomized, placebo-controlled, double-blind, phase II design performed for CoQ10 in 185 ALS patients, a decline in ALSFRS-R score and insufficient data were observed for further phase III testing [38].
2.4. Therapeutic Strategies against Glutamate-Induced Excitotoxicity
It has long been known that abnormal levels of glutamate, which is the main excitatory neurotransmitter in the brain, can cause neurodegenerative effects. The α-amino-3-hidroxy-5-methyl-4-isoxazole-propionic acid (AMPA) and the N-methyl-D-aspartate (NMDA) receptors, two important ionotropic glutamate receptors, are mediators of glutamate excitotoxicity owing to poor buffering calcium influx capacity of their subunits, such as the GluA2 subunit of the AMPA receptor. Massive entry of calcium into the cell stimulates phospholipases, proteases and endonucleases, causing devastation of energy metabolism and apoptotic or necrotic cell death. Besides, reduced expression of astrocytic excitatory amino-acid transporters (EAATs) is crucial for clearance of glutamate from the synaptic cleft into astrocytes, and defects in glutamate transport have been linked with ALS pathogenesis [39][40][41].
Riluzole (6-(trifluoromethoxy)-2-aminobenzothiazole) was discovered due to its remarkable anticonvulsant properties during the 1980s. The reaction of 4-(trifluoromethoxy)aniline with tetrabutylammonium thiocyanate afforded riluzole, in the presence of dichloromethane, with a yield of 61%. An alternative synthetic route for riluzole is the reaction of 4-(trifluoromethoxy)aniline with ammonium thiocyanate in the presence of acetonitrile (71%) (Figure 7) [42]. Riluzole (Figure 8) has been reported to exhibit neuroprotective effects in several animal models of PD, Huntington’s disease and cerebral ischemia [43]. Moreover, two large randomized placebo-controlled clinical trials were carried out to investigate the potency of riluzole in ALS treatment [44]. A double-blind, placebo-controlled phase II trial enrolling 155 ALS outpatients showed that riluzole slowed the disease progression and improve survival in patients [45]. In order to confirm and extend the therapeutic effect of riluzole, a double-blind, placebo-controlled, multicentre, international, dose-ranging study was performed on a large number of ALS outpatients. Results showed that it was well-tolerated and diminished the risk of death or tracheostomy in ALS patients [46]. Riluzole was approved by the FDA in 1995 and subsequently launched into the market under the trade name, Rilutek®. Riluzole, the first FDA-approved drug for the treatment of ALS, showed its mechanism via enhancement of extracellular glutamate uptake and inhibition of glutamate release from presynaptic terminals. Furthermore, riluzole also stabilizes the inactivated state of voltage-dependent sodium channels and NMDA receptor-mediated responses [47]. On the other hand, riluzole, dexpramipexole and pramipexole are 1,3-benzothiazole-based derivatives, though they display their anti-ALS activities in a different manner, implying the importance of the compounds carrying the same structural moiety endowed with different functions [48].
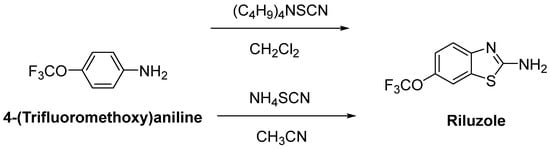
Figure 7. One-pot synthetic routes for riluzole.
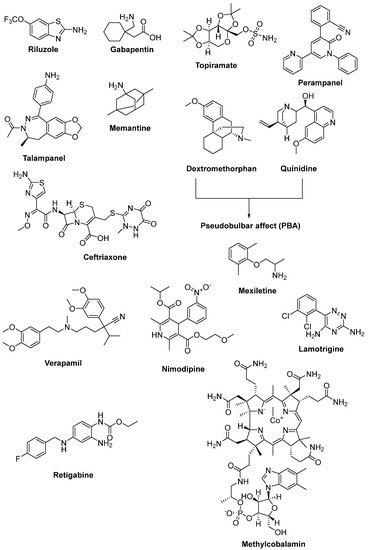
Figure 8. Therapeutics against glutamate-induced excitotoxicity.
2.5. Therapeutic Strategies for Reducing Apoptosis and/or Boosting Autophagy
Apoptosis is another potential mechanism of motor neuron death in ALS. It is termed as suicidal death of cells with a certain morphology, whereas autophagy is lysozyme-mediated sequestration of cytoplasmic material in vacuoles for bulk degradation. Autophagy is responsible for removing protein aggregates, damaged or redundant organelles through mitochondria or the endoplasmic reticulum along with toxic metabolites, cancer cells and intact microorganisms. Due to their lack of proliferation capacity and their high energy consumption, autophagy is essential for neurons to remove protein aggregates and damaged cellular inclusions. The Bcl-2 family proteins, as suppressors (Bcl-2, Bcl-XL) or promoters (Bax, Bad, Bak and Bcl-xS), regulate apoptosis, and it has recently become clear that they also control autophagy [49][50].
The Abelson non-receptor tyrosine kinase (c-Abl, Abl1) is normally activated in the Bcr–Abl hybrid protein of chronic myelogenous leukemia (CML). In addition to its oncogenic potential, overexpression of active c-Abl induces apoptosis and cell cycle arrest in response to a wide range of stimuli, such as inflammation, DNA damage, amyloid-β and oxidative stress, resulting in neurodegeneration and neuroinflammation [51][52]. It was also rationally hypothesized that activation of the Src/c-Abl signalling pathway synergistically leads to apoptosis in non-dividing cells (motor neurons) and the secretion of inflammatory factors in dividing cells (astrocytes and microglia) (Figure 9) [53]. There is growing evidence that the c-Abl pathway is a therapeutic target in ALS, based on the studies in cell culture revealing that inhibition of c-Abl protects cortical neurons from DNA damage-induced apoptosis [54]. Moreover, a robust rise in the phosphorylation of c-Abl in the brain and spinal cord of symptomatic SOD1G93A mice and in the spinal cord and motor cortex of symptomatic SOD1G86R mice was observed [55]. It was also documented that c-Abl expression was increased three-fold in post-mortem spinal cord tissues from sALS patients compared with non-ALS patients [56].
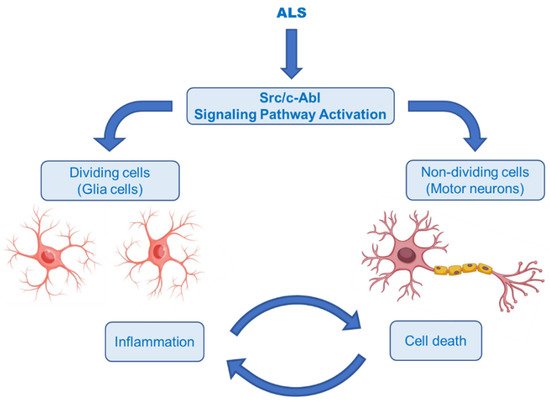
Katsumata et al. [56] designated that mutations of SOD1 activated the upregulation of c-Abl and alleviated cell viability. Additionally, dasatinib (Figure 10), a BBB-permeable c-Abl inhibitor, prevented cytotoxicity of mutant SOD1 proteins. Activation of c-Abl and caspase-3 was observed in SOD1G93A mice. Dasatinib extended the survival of SOD1G93A mice and diminished c-Abl phosphorylation. Imatinib (Figure 10), the c-Abl inhibitor used to treat CML, was reported to inhibit c-Abl phosphorylation in primary neuronal cultures at micromolar levels in response to various stimuli, such as oxidative stress. Imatinib was also determined to prevent astrocyte conditioned media (ACM)-hSOD1G93A-mediated motoneuron death [55]. Imamura et al. [58] determined that Src/c-Abl could be an efficient target for ALS treatment, based on HTS conducted for 1416 compounds, using ALS survival of motor neurons generated from an ALS patient harbouring SOD1 mutations. Bosutinib (Figure 10), a Src/c-Abl inhibitor used for treatment of CML, increased autophagy and reduced the levels of misfolded SOD1. Similar results were obtained with other mutations including TDP-43 and C9ORF72. Furthermore, bosutinib treatment modestly increased the life span of a mutant SOD1 ALS mice model. Bosutinib is currently in an open-label, multicentre phase I trial for ALS (UMIN000036295) to evaluate its safety and tolerability for the treatment of ALS patients and to explore its efficacy on ALS [59].
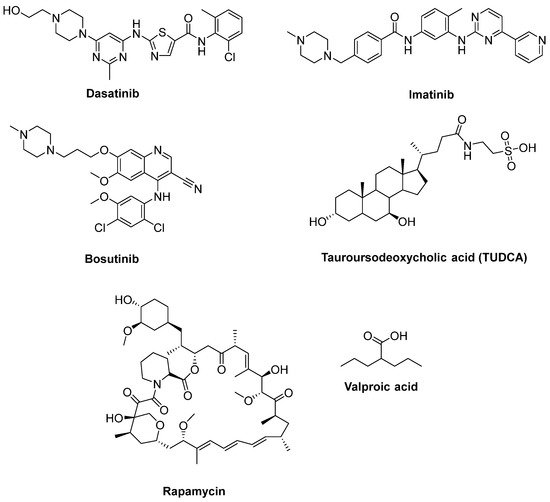
Figure 10. Therapeutics for reducing apoptosis and/or boosting autophagy.
2.6. Therapeutic Strategies against Neuroinflammation
Neuroinflammation is another important pathological mechanism in ALS progression. It is characterized by the activation of microglia and astrocytes, infiltration of immune cells (lymphocytes, macrophages, mast cells, neutrophils, etc.) and higher levels of inflammatory mediators. Microglia are generally divided into inflammatory (M1) and activated (M2) phenotypes. The M1 is linked with the release of proinflammatory cytokines (tumour necrosis factor (TNF)-α, interleukin-6 (IL-6), IL-23, IL-1β, IL-12, NO), cytotoxic substances (ROS), prostaglandin E2, chemokines, dysregulated glutamate levels, whereas M2 expresses anti-inflammatory molecules such as IL-10 and transforming growth factor (TGF)-β. There is mounting evidence regarding the connection between neuroinflammation with neuronal loss in sALS and fALS. Initially, glia and T cells, especially M2 macrophages/microglia, and T helper (Th) 2 cells and regulatory T (Treg) cells have protective roles in maintaining motor neuron viability. At a later stage, cytotoxic M1 macrophages/microglia and proinflammatory Th1 and Th17 T cells become more active [60][61][62][63][64].
Tocilizumab is an antibody that inhibits signalling of IL-6, a well-known promoter of the development of Th17 cells. It is currently used in patients with rheumatoid arthritis. A pilot study documented that in vitro tocilizumab mitigated several factors, including IL-6, that drive inflammation in sALS patients [65]. Another study including in vivo baseline inflammatory gene transcription in PBMCs of sALS patients indicated that tocilizumab infusions partially normalized inflammation of sALS patients [66]. A placebo-controlled phase II study with ALS patients who were genotyped for Asp358Ala polymorphism of the IL-6 receptor gene showed that tocilizumab treatment was safe and well-tolerated and alleviated C-reactive protein (CRP) levels in CSF relevant to IL-6 receptor Asp358Ala genotype. However, there was no difference in PBMC gene expression or clinical measures between groups [67].
Due to the efficacy of IL-1 inhibition in ALS, a single arm pilot study was performed for anakinra (Figure 11), an inhibitor of IL-1 receptor, in ALS patients. This study resulted in decreased levels of cytokines and the inflammatory marker fibrinogen during the first 24 weeks of treatment, but not in a significant reduction in disease progression [68].
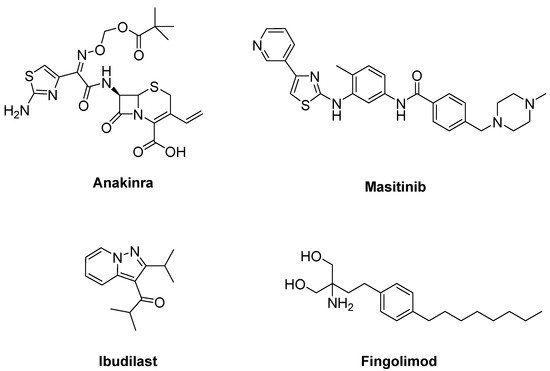
Figure 11. Therapeutics against neuroinflammation.
2.7. Therapeutic Strategies against Axonal Degeneration
The membrane protein Nogo-A is an inhibitor of neurite outgrowth that was initially identified as a potent myelin-associated inhibitor of axonal growth and regeneration [69]. Overexpressed, high amounts of Nogo-A were detected in skeletal muscles of ALS-linked mutant SOD1 mice and in patients with sALS [70]. Nogo-A causes retrograde axonal degeneration by destabilizing the neuromuscular junction in ALS [71]. Ozanezumab, an anti-Nogo-A monoclonal antibody, was found well-tolerated at single and repeat dose administration in a randomized, first-in-human clinical trial [72]. However, ozanezumab was not found to be potent, compared with placebo, in ALS patients in a double-blind, placebo-controlled, phase II trial [73].
Rho kinase (ROCK) plays an important role in the formation of high levels of actin filaments and a reduced actin turnover, leading to destruction of cell growth and axonal regeneration. Therefore, ROCK inhibition is a promising approach for ALS treatment [74]. Fasudil (Figure 12), an important ROCK inhibitor, was ascertained to slow disease progression, improve survival time and attenuate motor neuron loss in SOD1G93A mice [75]. A multicenter, double-blind, randomized, placebo-controlled phase IIa trial of fasudil in ALS patients also aims to assess the safety, tolerability and efficacy of fasudil at two different doses [76].
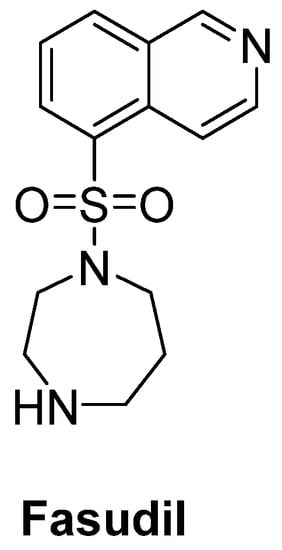
Figure 12. Fasudil, a promising ROCK inhibitor for alleviation of axonal degeneration.
2.8. Therapeutic Strategies against Skeletal Muscle Deterioration
Degenerated motor neurons result in progressive muscle atrophy in ALS. Fast skeletal muscle troponin activators (FSTA) such as tirasemtiv and reldesemtiv stimulate the troponin complex, increasing its susceptibility to calcium and modulate muscle contraction [27][71][77].
Tirasemtiv (Figure 13) was found to enhance rotarod performance in B6SJL-SOD1G93A transgenic mice with functional impairment [78]. In a randomized, double-blind, placebo-controlled trial on ALS patients, no treatment effect was ascertained for tirasemtiv, whereas slow vital capacity (SVC) and muscle strength declined at less than half the rate on tirasemtiv [79]. A phase III trial on ALS patients indicated that tirasemtiv showed no effect on the decline of SVC or secondary outcome measures. In spite of this unexpected outcome, the underlying mechanism of action was investigated with reldesemtiv (Figure 13), another fast skeletal muscle troponin activator [80]. A phase II, double-blind, randomized, dose-ranging trial on patients with ALS indicated that reldesemtiv was well-tolerated and stable against incremental rates of decline across multiple measures of ALS progression. For this reason, a phase III trial was planned for reldesemtiv [81].
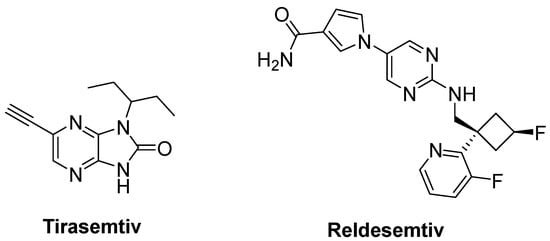
Figure 13. Therapeutics against skeletal muscle deterioration.
2.9. Therapeutic Strategies against Viruses
Apart from the aforementioned factors, new categories such as infectious agents, including viruses, bacteria and fungi, have been recently considered. Among these agents, there is mounting evidence reporting the connection between viruses and ALS pathogenesis. Increased nonspecific reverse transcriptase activity in the blood and CSF of ALS patients compared to relatives and controls also supports this connection. In particular, retroviruses such as human immunodeficiency virus (HIV) and human T-cell leukemia virus type-1 can cause ALS-like syndrome. Hence, antiretroviral therapy could be a potential approach for retarding the symptoms of the ALS-like syndrome associated with HIV infection [82][83][84][85].
A combination of nucleoside reverse-transcriptase inhibitors lamivudine and abacavir and an HIV-1 integrase strand transfer inhibitor dolutegravir (Triumeq®) (Figure 14) was considered to be effective in ALS treatment. An open-label phase IIa trial conducted in ALS patients revealed that long-term exposure to this combination was safe and well-tolerated in this cohort. A larger international phase III trial will be performed to investigate the effect of this combination on overall survival and ALS progression [86].
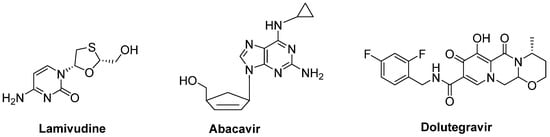
Figure 14. A potential combination of lamivudine, abacavir and dolutegravir to be effective in ALS.
2.10. The Importance of Trophic Factors in ALS
Trophic factors are essential for the enhancement of neuronal growth, survival and differentiation [87]. Growth factors are important for preventing motor neuron death in patients with ALS [88]. Due to its neurotrophic properties on motor neurons and the neuromuscular junction, recombinant human insulin-like growth factor I (rhIGF-I) was investigated for the determination of the relationship between rhIGF-I concentration and the prognosis of ALS. It was found that higher IGF-1 concentrations had the potential to increase survival [89]. Vascular endothelial growth factor (VEGF), a prominent factor for angiogenesis and neuroprotection, not only slowed the progression of ALS, but also increased life expectancy in SOD1G93A mice [90]. Another substantial neurotrophic factor, hepatocyte growth factor (HGF), alleviated motoneuron death and axonal degeneration and extended the life span of SOD1G93A mice [91]. A phase I study of HGF for ALS was performed from 2011 to 2015 at Tohoku University, Japan, and a phase II study commenced in May 2016 [92].
Intriguingly, epidermal growth factor receptor (EGFR) mRNA was detected to be overexpressed 10-fold more in the spinal cord of patients with ALS, just as in SOD1G93A transgenic mice model. Therefore, erlotinib (Figure 15), an important EGFR inhibitor, in particular for the treatment of EGFR mutated advanced or metastatic non-small-cell lung carcinoma (NSCLC), was tested on SOD1G93A mice. However, results exhibited that erlotinib could not extend survival, though it delayed the onset of multiple behavioural measures of disease progression [93].
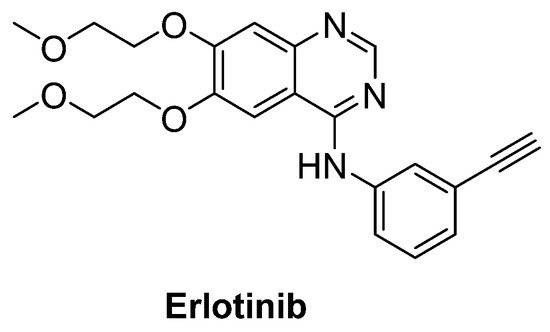
Figure 15. Erlotinib, an important EGFR inhibitor.
2.11. Newly Synthesized and Evaluated Compounds as Anti-ALS Agents
Chen et al. [94] defined two arylsulfanyl pyrazolone (ASP) bearing compounds, 5-((2,4-dichloro-5-methylphenylthio)methyl)-1H-pyrazol-3(2H)-one (1) and 5-((4-chloro-2,5-dimethylphenylthio)methyl)-1H-pyrazol-3(2H)-one (2) (Figure 16), as chemical hits by means of HTS assay expressing mutant SOD1G93A. Then, 5-((3,5-dichlorophenylthio)methyl)-1H-pyrazol-3(2H)-one (19) (Figure 16), which was generated based on the structural optimization of ASP scaffold, was found to be more potent, with an EC50 value of 170 nM. According to pharmacokinetic assays, general parameters were amenable for compound 1, except for its relatively rapid clearance and short microsomal half-life stability features. The optimization of ASP-based compounds stands out as novel therapeutic candidates for ALS treatment.
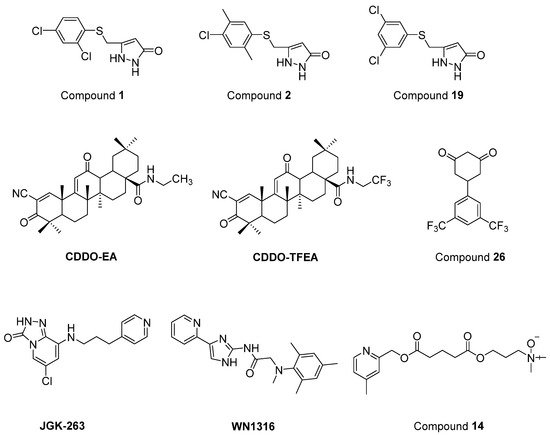
Figure 16. Newly synthesized and evaluated compounds as anti-ALS agents.
Nrf2/ARE (NF-E2-related factor 2/antioxidant response element) signalling program has been reported to be important in the regulation of oxidative damage, neuroinflammation and mitochondrial dysfunction. CDDO (2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid) ethylamide (CDDO-EA) and CDDO trifluoroethylamide (CDDO-TFEA) (Figure 16) are two synthetic triterpenoid analogs derived from oleanolic acid. It was determined that these two oleanolic acid derivatives stimulated Nrf2/ARE in NSC-34 cell culture and in the SOD1G93A ALS mouse model [95].
Cyclohexane-1,3-dione (CHD) derivatives were identified based on PC12-G93A-YFP HTS assay [96]. A more efficient compound with an EC50 value of 700 nM and more favourable pharmacokinetic profile, 5-(3,5-Bis(trifluoromethyl)phenyl)cyclohexane-1,3-dione (26) (Figure 16), was obtained via the structural modification of the CHD scaffold. However, compound 26 revealed no significant activity in the mutant SOD1G93A mice model. These CHD analogs could be novel therapeutic candidates for further ALS studies.
A new GSK-3β inhibitor, 6-chloro-8-((3-(pyridin-4-yl)propyl)amino)-[1][2][4]triazolo[4,3-a]pyridin-3(2H)-one (JGK-263) (Figure 16) was developed, and its improved viability and neuroprotection was determined in normal and SOD1 wild/mutant NSC34 cell lines [97]. It was also observed that JGK-263 improved motor function and delayed the time until symptom onset and death in SOD1G93A ALS mice model.
Tanaka et al. [98] documented that 2-[mesityl(methyl)amino]-N-[4-(pyridin-2-yl)-1H-imidazol-2-yl] acetamide trihydrochloride (WN1316) (Figure 16) selectively slowed oxidative stress-induced cell death and neuronal inflammation in the late-stage of ALS mice. WN1316, with high water solubility and BBB permeability, increased neuronal apoptosis inhibitory protein (NAIP) and Nrf2. Post-onset oral administration of low dose WN1316 improved motor function and extended the survival in SOD1H46R and SOD1G93A ALS mice models. The results of phase I clinical trial of WN1316 (UMIN000015054) have not been published yet.
Several ester and amide derivatives of chemical chaperones were synthesized by Getter et al. [99]. Among them, 3-((5-((4,6-dimethylpyridin-2-yl)methoxy)-5-oxopentanoyl)oxy)-N,N-dimethylpropan-1-amine oxide (14) (Figure 16) in vitro displayed both neuronal and astrocyte protection. Moreover, compound 14 improved the neurological functions and prolonged body weight loss in ALS mice.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23052400
References
- Liu, J.; Wang, F. Role of neuroinflammation in amyotrophic lateral sclerosis: Cellular mechanisms and therapeutic implications. Front. Immunol. 2017, 8, 1005.
- Martinez, A.; Palomo Ruiz, M.D.; Perez, D.I.; Gil, C. Drugs in clinical development for the treatment of amyotrophic lateral sclerosis. Expert Opin. Investig. Drugs 2017, 26, 403–414.
- Gordon, P.H. Amyotrophic lateral sclerosis: Pathophysiology, diagnosis and management. CNS Drugs 2011, 25, 1–15.
- Harikrishnareddy, D.; Misra, S.; Upadhyay, S.; Modi, M.; Medhi, B. Roots to start research in amyotrophic lateral sclerosis: Molecular pathways and novel therapeutics for future. Rev. Neurosci. 2015, 26, 161–181.
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641.
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative stress, neuroinflammation and mitochondria in the pathophysiology of amyotrophic lateral sclerosis. Antioxidants 2020, 9, 901.
- Harley, J.; Clarke, B.E.; Patani, R. The interplay of RNA binding proteins, oxidative stress and mitochondrial dysfunction in ALS. Antioxidants 2021, 10, 552.
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The link between oxidative stress, redox status, bioenergetics and mitochondria in the pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352.
- Watanabe, K.; Morinaka, Y.; Iseki, K.; Watanabe, T.; Yuki, S.; Nishi, H. Structure-activity relationship of 3-methyl-1-phenyl-2-pyrazolin-5-one (edaravone). Redox Rep. 2003, 8, 151–155.
- Watanabe, K.; Tanaka, M.; Yuki, S.; Hirai, M.; Yamamoto, Y. How is edaravone effective against acute ischemic stroke and amyotrophic lateral sclerosis? J. Clin. Biochem. Nutr. 2018, 62, 20–38.
- Nakagawa, H.; Ohyama, R.; Kimata, A.; Suzuki, T.; Miyata, N. Hydroxyl radical scavenging by edaravone derivatives: Efficient scavenging by 3-methyl-1-(pyridin-2-yl)-5-pyrazolone with an intramolecular base. Bioorg. Med. Chem. Lett. 2006, 16, 5939–5942.
- Nayak, M.; Batchu, H.; Batra, S. Straightforward copper-catalyzed synthesis of pyrrolopyrazoles from halogenated pyrazolecarbaldehydes. Tetrahedron Lett. 2012, 53, 4206–4208.
- Vijay, K.; Nandi, C.; Samant, S.D. Synthesis of a dihydroquinoline based merocyanine as a ‘naked eye’ and ‘fluorogenic’ sensor for hydrazine hydrate in aqueous medium and hydrazine gas. RSC Adv. 2014, 4, 30712–30717.
- Yoshino, H.; Kimura, A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph. Lateral Scler. 2006, 7, 241–245.
- Writing Group; Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512.
- Takei, K.; Watanabe, K.; Yuki, S.; Akimoto, M.; Sakata, T.; Palumbo, J. Edaravone and its clinical development for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 5–10.
- Dash, R.P.; Babu, R.J.; Srinivas, N.R. Two decades-long journey from riluzole to edaravone: Revisiting the clinical pharmacokinetics of the only two amyotrophic lateral sclerosis therapeutics. Clin. Pharmacokinet. 2018, 57, 1385–1398.
- Bailly, C.; Hecquet, P.E.; Kouach, M.; Thuru, X.; Goossens, J.F. Chemical reactivity and uses of 1-phenyl-3-methyl-5-pyrazolone (PMP), also known as edaravone. Bioorg. Med. Chem. 2020, 28, 115463.
- Choi, E.S.; Dokholyan, N.V. SOD1 oligomers in amyotrophic lateral sclerosis. Curr. Opin. Struct. Biol. 2021, 66, 225–230.
- Eleutherio, E.C.A.; Silva Magalhães, R.S.; de Araújo Brasil, A.; Monteiro Neto, J.R.; de Holanda Paranhos, L. SOD1, more than just an antioxidant. Arch. Biochem. Biophys. 2021, 697, 108701.
- Smith, R.A.; Milleri, T.M.; Yamanaka, K.; Monia, B.P.; Condon, T.P.; Hung, G.; Lobsiger, C.S.; Ward, C.M.; McAlonis-Downes, M.; Wei, H.; et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Investig. 2006, 116, 2290–2296.
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 2013, 12, 435–442.
- Miller, T.; Cudkowitz, M.; Shaw, P. Safety, PK, PD, and exploratory efficacy in single and multiple dose study of a SOD1 antisense oligonucleotide (BIIB067) administered to participants with ALS. In Proceedings of the American Academy of Neurology 71st Annual Meeting, Philadelphia, PA, USA, 4–10 May 2019.
- Kieran, D.; Kalmar, B.; Dick, J.R.; Riddoch-Contreras, J.; Burnstock, G.; Greensmith, L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat. Med. 2004, 10, 402–405.
- Kalmar, B.; Lu, C.H.; Greensmith, L. The role of heat shock proteins in Amyotrophic Lateral Sclerosis: The therapeutic potential of Arimoclomol. Pharmacol. Ther. 2014, 141, 40–54.
- Benatar, M.; Wuu, J.; Andersen, P.M.; Atassi, N.; David, W.; Cudkowicz, M.; Schoenfeld, D. Randomized, double-blind, placebo-controlled trial of arimoclomol in rapidly progressive SOD1 ALS. Neurology 2018, 90, e565–e574.
- Liscic, R.M.; Alberici, A.; Cairns, N.J.; Romano, M.; Buratti, E. From basic research to the clinic: Innovative therapies for ALS and FTD in the pipeline. Mol. Neurodegener. 2020, 15, 31.
- Crippa, V.; D’Agostino, V.G.; Cristofani, R.; Rusmini, P.; Cicardi, M.E.; Messi, E.; Loffredo, R.; Pancher, M.; Piccolella, M.; Galbiati, M.; et al. Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci. Rep. 2016, 6, 22827.
- Mandrioli, J.; Crippa, V.; Cereda, C.; Bonetto, V.; Zucchi, E.; Gessani, A.; Ceroni, M.; Chio, A.; D’Amico, R.; Monsurrò, M.R.; et al. Proteostasis and ALS: Protocol for a phase II, randomised, double-blind, placebo-controlled, multicentre clinical trial for colchicine in ALS (Co-ALS). BMJ Open. 2019, 9, e028486.
- Jhanji, R.; Behl, T.; Sehgal, A.; Bungau, S. Mitochondrial dysfunction and traffic jams in amyotrophic lateral sclerosis. Mitochondrion 2021, 58, 102–110.
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galéa, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A.; et al. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720.
- Lenglet, T.; Lacomblez, L.; Abitbol, J.L.; Ludolph, A.; Mora, J.S.; Robberecht, W.; Shaw, P.J.; Pruss, R.M.; Cuvier, V.; Meininger, V.; et al. A phase II-III trial of olesoxime in subjects with amyotrophic lateral sclerosis. Eur. J. Neurol. 2014, 21, 529–536.
- Klivenyi, P.; Ferrante, R.J.; Matthews, R.T.; Bogdanov, M.B.; Klein, A.M.; Andreassen, O.A.; Mueller, G.; Wermer, M.; Kaddurah-Daouk, R.; Beal, M.F. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat. Med. 1999, 5, 347–350.
- Groeneveld, G.J.; Veldink, J.H.; van der Tweel, I.; Kalmijn, S.; Beijer, C.; de Visser, M.; Wokke, J.H.; Franssen, H.; van den Berg, L.H. A randomized sequential trial of creatine in amyotrophic lateral sclerosis. Ann. Neurol. 2003, 53, 437–445.
- Shefner, J.M.; Cudkowicz, M.E.; Schoenfeld, D.; Conrad, T.; Taft, J.; Chilton, M.; Urbinelli, L.; Qureshi, M.; Zhang, H.; Pestronk, A.; et al. A clinical trial of creatine in ALS. Neurology 2004, 63, 1656–1661.
- Rosenfeld, J.; King, R.M.; Jackson, C.E.; Bedlack, R.S.; Barohn, R.J.; Dick, A.; Phillips, L.H.; Chapin, J.; Gelinas, D.F.; Lou, J.S. Creatine monohydrate in ALS: Effects on strength, fatigue, respiratory status and ALSFRS. Amyotroph. Lateral Scler. 2008, 9, 266–272.
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897.
- Kaufmann, P.; Thompson, J.L.; Levy, G.; Buchsbaum, R.; Shefner, J.; Krivickas, L.S.; Katz, J.; Rollins, Y.; Barohn, R.J.; Jackson, C.E.; et al. Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann. Neurol. 2009, 66, 235–244.
- Ramalho, T.C.; de Castro, A.A.; Tavares, T.S.; Silva, M.C.; Silva, D.R.; Cesar, P.H.; Santos, L.A.; da Cunha, E.F.F.; Nepovimova, E.; Kuca, K. Insights into the pharmaceuticals and mechanisms of neurological orphan diseases: Current status and future expectations. Prog. Neurobiol. 2018, 169, 135–157.
- Ng Kee Kwong, K.C.; Mehta, A.R.; Nedergaard, M.; Chandran, S. Defining novel functions for cerebrospinal fluid in ALS pathophysiology. Acta Neuropathol. Commun. 2020, 8, 140.
- Silva-Hucha, S.; Pastor, A.M.; Morcuende, S. Neuroprotective effect of vascular endothelial growth factor on motoneurons of the oculomotor system. Int. J. Mol. Sci. 2021, 22, 814.
- Cheah, B.C.; Vucic, S.; Krishnan, A.V.; Kiernan, M.C. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr. Med. Chem. 2010, 17, 1942–1959.
- Mignani, S.; Majoral, J.P.; Desaphy, J.F.; Lentini, G. From Riluzole to dexpramipexole via substituted-benzothiazole derivatives for amyotrophic lateral sclerosis disease treatment: Case studies. Molecules 2020, 25, 3320.
- Mathis, S.; Couratier, P.; Julian, A.; Vallat, J.M.; Corcia, P.; Le Masson, G. Management and therapeutic perspectives in amyotrophic lateral sclerosis. Expert Rev. Neurother. 2017, 17, 263–276.
- Bensimon, G.; Lacomblez, L.; Meininger, V. ALS/Riluzole Study Group. A controlled trial of riluzole in amyotrophic lateral sclerosis. N. Engl. J. Med. 1994, 330, 585–591.
- Lacomblez, L.; Bensimon, G.; Leigh, P.N.; Guillet, P.; Powe, L.; Durrleman, S.; Delumeau, J.C.; Meininger, V. A confirmatory dose-ranging study of riluzole in ALS. ALS/Riluzole Study Group-II. Neurology 1996, 47, S242–S250.
- Dorst, J.; Ludolph, A.C.; Huebers, A. Disease-modifying and symptomatic treatment of amyotrophic lateral sclerosis. Ther. Adv. Neurol. Disord. 2017, 11, 1756285617734734.
- Kumar, V.; Islam, A.; Hassan, M.I.; Ahmad, F. Therapeutic progress in amyotrophic lateral sclerosis-beginning to learning. Eur. J. Med. Chem. 2016, 121, 903–917.
- Levine, B.; Sinha, S.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606.
- Amin, A.; Perera, N.D.; Beart, P.M.; Turner, B.J.; Shabanpoor, F. Amyotrophic lateral sclerosis and autophagy: Dysfunction and therapeutic targeting. Cells 2020, 9, 2413.
- Wang, J. Regulation of cell death by the Abl tyrosine kinase. Oncogene 2000, 19, 5643–5650.
- Schlatterer, S.D.; Acker, C.M.; Davies, P. c-Abl in neurodegenerative disease. J. Mol. Neurosci. 2011, 45, 445–452.
- Karagiannis, P.; Inoue, H. ALS, a cellular whodunit on motor neuron degeneration. Mol. Cell Neurosci. 2020, 107, 103524.
- Kim, B.W.; Jeong, Y.E.; Wong, M.; Martin, L.J. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol. Commun. 2020, 8, 7.
- Rojas, F.; Gonzalez, D.; Cortes, N.; Ampuero, E.; Hernández, D.E.; Fritz, E.; Abarzua, S.; Martinez, A.; Elorza, A.A.; Alvarez, A.; et al. Reactive oxygen species trigger motoneuron death in non-cell-autonomous models of ALS through activation of c-Abl signaling. Front. Cell Neurosci. 2015, 9, 203.
- Katsumata, R.; Ishigaki, S.; Katsuno, M.; Kawai, K.; Sone, J.; Huang, Z.; Adachi, H.; Tanaka, F.; Urano, F.; Sobue, G. c-Abl inhibition delays motor neuron degeneration in the G93A mouse, an animal model of amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e46185.
- Smart Servier Medical Art. Available online: https://smart.servier.com/ (accessed on 4 November 2021).
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962.
- Imamura, K.; Izumi, Y.; Banno, H.; Uozumi, R.; Morita, S.; Egawa, N.; Ayaki, T.; Nagai, M.; Nishiyama, K.; Watanabe, Y.; et al. Induced pluripotent stem cell-based Drug Repurposing for Amyotrophic lateral sclerosis Medicine (iDReAM) study: Protocol for a phase I dose escalation study of bosutinib for amyotrophic lateral sclerosis patients. BMJ Open 2019, 9, e033131.
- Freeman, L.C.; Ting, J.P. The pathogenic role of the inflammasome in neurodegenerative diseases. J. Neurochem. 2016, 136, 29–38.
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 1181–1194.
- Du, L.; Zhang, Y.; Chen, Y.; Zhu, J.; Yang, Y.; Zhang, H.L. Role of microglia in neurological disorders and their potentials as a therapeutic target. Mol. Neurobiol. 2017, 54, 7567–7584.
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The dual role of microglia in ALS: Mechanisms and therapeutic approaches. Front. Aging Neurosci. 2017, 9, 242.
- Kulczyńska-Przybik, A.; Mroczko, P.; Dulewicz, M.; Mroczko, B. The implication of reticulons (RTNs) in neurodegenerative diseases: From molecular mechanisms to potential diagnostic and therapeutic approaches. Int. J. Mol. Sci. 2021, 22, 4630.
- Mizwicki, M.T.; Fiala, M.; Magpantay, L.; Aziz, N.; Sayre, J.; Liu, G.; Siani, A.; Chan, D.; Martinez-Maza, O.; Chattopadhyay, M.; et al. Tocilizumab attenuates inflammation in ALS patients through inhibition of IL6 receptor signaling. Am. J. Neurodegener. Dis. 2012, 1, 305–315.
- Fiala, M.; Mizwicki, M.T.; Weitzman, R.; Magpantay, L.; Nishimoto, N. Tocilizumab infusion therapy normalizes inflammation in sporadic ALS patients. Am. J. Neurodegener. Dis. 2013, 2, 129–139.
- Milligan, C.; Atassi, N.; Babu, S.; Barohn, R.J.; Caress, J.B.; Cudkowicz, M.E.; Evora, A.; Hawkins, G.A.; Wosiski-Kuhn, M.; Macklin, E.A.; et al. Tocilizumab is safe and tolerable and reduces C-reactive protein concentrations in the plasma and cerebrospinal fluid of ALS patients. Muscle Nerve 2021, 64, 309–320.
- Maier, A.; Deigendesch, N.; Müller, K.; Weishaupt, J.H.; Krannich, A.; Röhle, R.; Meissner, F.; Molawi, K.; Münch, C.; Holm, T.; et al. Interleukin-1 antagonist anakinra in amyotrophic lateral sclerosis-A Pilot Study. PLoS ONE 2015, 10, e0139684.
- Lynch, A.M.; Clevel, M.; Prinjha, R.; Kumar, U.; Stubbs, R.; Wuerthner, J. Non-clinical development of ozanezumab: A humanised antibody targeting the amino terminus of neurite outgrowth inhibitor A (Nogo-A). Toxicol. Res. 2015, 4, 1333.
- Jokic, N.; Gonzalez de Aguilar, J.L.; Dimou, L.; Lin, S.; Fergani, A.; Ruegg, M.A.; Schwab, M.E.; Dupuis, L.; Loeffler, J.P. The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model. EMBO Rep. 2006, 7, 1162–1167.
- Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C. Skeletal Muscle in ALS: An unappreciated therapeutic opportunity? Cells 2021, 10, 525.
- Meininger, V.; Pradat, P.F.; Corse, A.; Al-Sarraj, S.; Rix Brooks, B.; Caress, J.B.; Cudkowicz, M.; Kolb, S.J.; Lange, D.; Leigh, P.N.; et al. Safety, pharmacokinetic, and functional effects of the Nogo-A monoclonal antibody in amyotrophic lateral sclerosis: A randomized, first-in-human clinical trial. PLoS ONE 2014, 9, e97803.
- Meininger, V.; Genge, A.; van den Berg, L.H.; Robberecht, W.; Ludolph, A.; Chio, A.; Kim, S.H.; Leigh, P.N.; Kiernan, M.C.; Shefner, J.M.; et al. Safety and efficacy of ozanezumab in patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2017, 16, 208–216.
- Koch, J.C.; Kuttler, J.; Maass, F.; Lengenfeld, T.; Zielke, E.; Bähr, M.; Lingor, P. Compassionate use of the ROCK inhibitor fasudil in three patients with amyotrophic lateral sclerosis. Front. Neurol. 2020, 11, 173.
- Takata, M.; Tanaka, H.; Kimura, M.; Nagahara, Y.; Tanaka, K.; Kawasaki, K.; Seto, M.; Tsuruma, K.; Shimazawa, M.; Hara, H. Fasudil, a rho kinase inhibitor, limits motor neuron loss in experimental models of amyotrophic lateral sclerosis. Br. J. Pharmacol. 2013, 170, 341–351.
- Lingor, P.; Weber, M.; Camu, W.; Friede, T.; Hilgers, R.; Leha, A.; Neuwirth, C.; Günther, R.; Benatar, M.; Kuzma-Kozakiewicz, M.; et al. ROCK-ALS: Protocol for a randomized, placebo-controlled, double-blind phase IIa trial of safety, tolerability and efficacy of the rho kinase (ROCK) inhibitor fasudil in amyotrophic lateral sclerosis. Front. Neurol. 2019, 10, 293.
- Wobst, H.J.; Mack, K.L.; Brown, D.G.; Brandon, N.J.; Shorter, J. The clinical trial landscape in amyotrophic lateral sclerosis—Past, present, and future. Med. Res. Rev. 2020, 40, 1352–1384.
- Hwee, D.T.; Kennedy, A.; Ryans, J.; Russell, A.J.; Jia, Z.; Hinken, A.C.; Morgans, D.J.; Malik, F.I.; Jasper, J.R. Fast skeletal muscle troponin activator tirasemtiv increases muscle function and performance in the B6SJL-SOD1G93A ALS mouse model. PLoS ONE 2014, 9, e96921.
- Shefner, J.M.; Wolff, A.A.; Meng, L.; Bian, A.; Lee, J.; Barragan, D.; Andrews, J.A. BENEFIT-ALS Study Group. A randomized, placebo-controlled, double-blind phase IIb trial evaluating the safety and efficacy of tirasemtiv in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 426–435.
- Andrews, J.A.; Cudkowicz, M.E.; Hardiman, O.; Meng, L.; Bian, A.; Lee, J.; Wolff, A.A.; Malik, F.I.; Shefner, J.M. VITALITY-ALS, a phase III trial of tirasemtiv, a selective fast skeletal muscle troponin activator, as a potential treatment for patients with amyotrophic lateral sclerosis: Study design and baseline characteristics. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 259–266.
- Shefner, J.M.; Andrews, J.A.; Genge, A.; Jackson, C.; Lechtzin, N.; Miller, T.M.; Cockroft, B.M.; Meng, L.; Wei, J.; Wolff, A.A.; et al. A phase 2, double-blind, randomized, dose-ranging trial of reldesemtiv in patients with ALS. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 287–299.
- MacGowan, D.J.; Scelsa, S.N.; Imperato, T.E.; Liu, K.N.; Baron, P.; Polsky, B. A controlled study of reverse transcriptase in serum and CSF of HIV-negative patients with ALS. Neurology 2007, 68, 1944–1946.
- McCormick, A.L.; Brown, R.H., Jr.; Cudkowicz, M.E.; Al-Chalabi, A.; Garson, J.A. Quantification of reverse transcriptase in ALS and elimination of a novel retroviral candidate. Neurology 2008, 70, 278–283.
- Douville, R.; Liu, J.; Rothstein, J.; Nath, A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann. Neurol. 2011, 69, 141–151.
- Castanedo-Vazquez, D.; Bosque-Varela, P.; Sainz-Pelayo, A.; Riancho, J. Infectious agents and amyotrophic lateral sclerosis: Another piece of the puzzle of motor neuron degeneration. J. Neurol. 2019, 266, 27–36.
- Gold, J.; Rowe, D.B.; Kiernan, M.C.; Vucic, S.; Mathers, S.; van Eijk, R.P.A.; Nath, A.; Garcia Montojo, M.; Norato, G.; Santamaria, U.A.; et al. Safety and tolerability of Triumeq in amyotrophic lateral sclerosis: The lighthouse trial. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 595–604.
- Nagtegaal, I.D.; Lakke, E.A.; Marani, E. Trophic and tropic factors in the development of the central nervous system. Arch. Physiol. Biochem. 1998, 106, 161–202.
- Zinman, L.; Cudkowicz, M. Emerging targets and treatments in amyotrophic lateral sclerosis. Lancet Neurol. 2011, 10, 481–490.
- Nagel, G.; Peter, R.S.; Rosenbohm, A.; Koenig, W.; Dupuis, L.; Rothenbacher, D.; Ludolph, A.C. Association of Insulin-like Growth Factor 1 Concentrations with Risk for and Prognosis of Amyotrophic Lateral Sclerosis—Results from the ALS Registry Swabia. Sci. Rep. 2020, 10, 736.
- Azzouz, M.; Ralph, G.S.; Storkebaum, E.; Walmsley, L.E.; Mitrophanous, K.A.; Kingsman, S.M.; Carmeliet, P.; Mazarakis, N.D. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 2004, 429, 413–417.
- Sun, W.; Funakoshi, H.; Nakamura, T. Overexpression of HGF retards disease progression and prolongs life span in a transgenic mouse model of ALS. J. Neurosci. 2002, 22, 6537–6548.
- Kitamura, K.; Nagoshi, N.; Tsuji, O.; Matsumoto, M.; Okano, H.; Nakamura, M. Application of hepatocyte growth factor for acute spinal cord injury: The road from basic studies to human treatment. Int. J. Mol. Sci. 2019, 20, 1054.
- Le Pichon, C.E.; Dominguez, S.L.; Solanoy, H.; Ngu, H.; Lewin-Koh, N.; Chen, M.; Eastham-Anderson, J.; Watts, R.; Scearce-Levie, K. EGFR inhibitor erlotinib delays disease progression but does not extend survival in the SOD1 mouse model of ALS. PLoS ONE 2013, 8, e62342.
- Chen, T.; Benmohamed, R.; Arvanites, A.C.; Ralay Ranaivo, H.; Morimoto, R.I.; Ferrante, R.J.; Watterson, D.M.; Kirsch, D.R.; Silverman, R.B. Arylsulfanyl pyrazolones block mutant SOD1-G93A aggregation. Potential application for the treatment of amyotrophic lateral sclerosis. Bioorg. Med. Chem. 2011, 19, 613–622.
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingsong, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96.
- Zhang, W.; Benmohamed, R.; Arvanites, A.C.; Morimoto, R.I.; Ferrante, R.J.; Kirsch, D.R.; Silverman, R.B. Cyclohexane 1,3-diones and their inhibition of mutant SOD1-dependent protein aggregation and toxicity in PC12 cells. Bioorg. Med. Chem. 2012, 20, 1029–1045.
- Ahn, S.W.; Jeon, G.S.; Kim, M.J.; Shon, J.H.; Kim, J.E.; Shin, J.Y.; Kim, S.M.; Kim, S.H.; Ye, I.H.; Lee, K.W.; et al. Neuroprotective effects of JGK-263 in transgenic SOD1-G93A mice of amyotrophic lateral sclerosis. J. Neurol. Sci. 2014, 340, 112–116.
- Tanaka, K.; Kanno, T.; Yanagisawa, Y.; Yasutake, K.; Inoue, S.; Hirayama, N.; Ikeda, J.E. A novel acylaminoimidazole derivative, WN1316, alleviates disease progression via suppression of glial inflammation in ALS mouse model. PLoS ONE 2014, 9, e87728.
- Getter, T.; Zaks, I.; Barhum, Y.; Ben-Zur, T.; Böselt, S.; Gregoire, S.; Viskind, O.; Shani, T.; Gottlieb, H.; Green, O.; et al. A chemical chaperone-based drug candidate is effective in a mouse model of amyotrophic lateral sclerosis (ALS). ChemMedChem 2015, 10, 850–861.
This entry is offline, you can click here to edit this entry!