2.1. The Signaling Pathways That Activate Cell Cycle Checkpoint Response
The cytotoxicity caused by IR is mainly the result of DNA damage. IR induces several forms of DNA damage, which include single-stranded DNA breaks (SSBs), double-strand DNA breaks (DSBs), sugar and base damages, and DNA-protein crosslinks [
12,
17,
18]. Among those, DSBs are the most lethal form of DNA damage, as misrepaired and unrepaired DSBs will result in genomic instability and cell death, respectively [
19,
20].
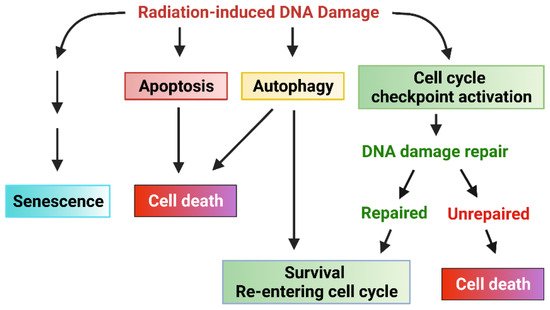
Upon DNA damage being sensed following IR, the cell cycle checkpoint will be activated to allow time for DNA repair [
21]. Three cell cycle checkpoints exist, termed the G1 checkpoint, intra-S checkpoint, and G2 checkpoint, which block the cell cycle progression at the G1/S border, intra-S, or G2/M border, respectively, after detecting the DNA damage [
21]. If the cell cycle checkpoints are defective in the cells or the DNA damage cannot be unrepaired, other responses (such as apoptosis, senescence, autophagy cell death, or necrosis) may be activated to eliminate the injured cells [
21]. Therefore, a properly functional cell cycle checkpoint facilitates DNA repair in cancer cells, which is anticipated to promote the cell survival in response to IR.
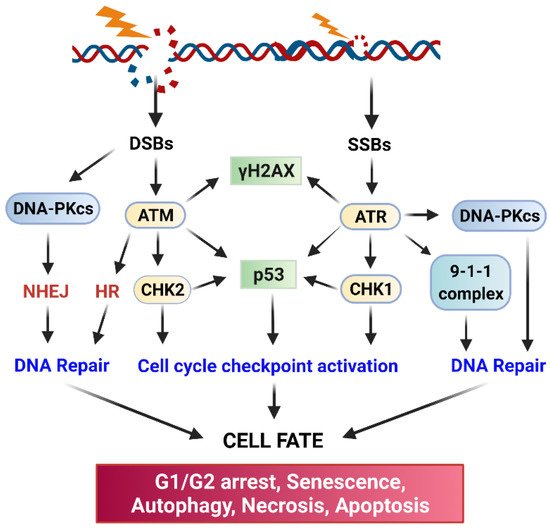
ATM (Ataxia Telangiesctasia Mutated) and ATR (Ataxia Telangiectasia and Rad3-related) kinase-mediated signaling pathways play essential roles in the activation of cell cycle checkpoint response and DNA repair following radiation-induced DNA damage (
Figure 2) [
21,
22]. Upon sensing DNA damage, ATM and ATR are rapidly activated, which, in turn, activate their downstream targets, including p53, DNA-activated protein kinase (DNA-PK), Checkpoint kinase (Chk)1, and Chk2 [
21,
22]. Activation of the Chk1/2 kinases results in the phosphorylation of Cell division control protein (Cdc)25 phosphatase, which leads to the subcellular sequestration (by 14-3-3), degradation, and/or inhibition of Cdc25 that otherwise activate the Cdk1 (Cyclin-dependent kinase 1)/Cyclin B activity to promote the G2/M transition of the cell cycle [
23]. Furthermore, in response to IR, ATM, and ATR kinases, as well as Chk1 and Chk2 kinases, can directly phosphorylate and activate p53 tumor suppressor [
21,
22,
24]. Consequently, activation of p53 by ATM, ATR, Chk1, and Chk2 results in a marked induction of p21 protein, which directly inhibits the activities of the Cdk4/Cyclin D, Cdk6/Cyclin D, and Cdk1/Cyclin A/B complexes to block the cell cycle progression [
21,
22].
Cell cycle progression requires the activities of Cdk kinases. While the G1/S transition of the cycle requires the activity of Cdk4/Cdk6 coupled with Cyclin D, the G2/M transition of the cell cycle requires the activity of Cdk1 coupled with Cyclin B [
25,
26]. The G1 checkpoint is mainly guarded by the p53 tumor suppressor and its transcriptional target p21, which directly binds to and inhibits Cdk4/6 [
27]. The G2 checkpoint is controlled by the Cdk1/Cyclin B complex [
26]. It is known that most cancer cells are defective in the G1 checkpoint due to the common mutations in the key regulators of the G1 checkpoint (e.g., p53, Cyclin D) [
27]. However, most cancer cells possess a functional G2 checkpoint, which is operated mainly through p53-independent mechanisms [
28]. Thus, abrogation of the G2 checkpoint in the cancer cells that are defective in the G1 checkpoint can sensitize the cells to radiation [
29].
The inhibitory phosphorylation of Cdk1-Y15 by the Wee1 and Myt1 kinases inhibits Cdk1 activity, and it is the essential step for the activation of the G2 checkpoint by radiation [
30]. Cdk1-Y15 resides in the ATP-binding domain of Cdk1, and phosphorylation of this site prevents the binding of ATP to Cdk1, thus inhibiting Cdk1 activity. The dephosphorylation of Cdk1-Y15 is catalyzed by the Cdc25 dual-specificity phosphatase that activates the Cdk1 activity [
31,
32,
33]. During IR-induced G2/M cell cycle arrest, the phosphorylation of Cdk1-Y15 is maintained [
30,
34,
35].
ATM, ATR, and DNA-PK also serve as major activators of DNA repair, and each of them is recruited to the DNA damage sites by a specific co-factor: Nijmegen breakage syndrome 1 (NBS1) (a component of the MRE11-RAD50-NBS1 complex) for ATM [
36,
37,
38], ATRIP for ATR [
39], and Ku80 for DNA-PKcs [
40,
41]. The initiation step will trigger the subsequent recruitment of additional co-factors required for the assembly of DNA repair apparatus at the sites of DNA damage. biochemically, DNA-PK primarily triggers DSB repair via non-homologous end-joining repair (NHEJ), ATM triggers both NHEJ and homologous recombination (HR) repair of DSB, and ATR mainly triggers HR-mediated DSB repair [
22,
42]. In addition, there is functional redundancy and crosstalk among the three DNA damage response (DDR) pathways. Ultimately, activation of the three pathways by radiation results in the inhibition of Cdk activities leading to cell cycle arrest to allow time for DNA repair and cell survival [
12].
2.2. DNA Repair Pathways
In response to the DNA damage by IR, cancer cells rapidly activate ATM, ATR, and DNA-PK, all of which are members of the phosphoinositide 3 kinase-related kinase family. These kinases transduce the DNA damage signaling, coordinate the assembly of DNA repairing apparatuses at the damaged sites and initiate the repairing of DNA (
Figure 2) [
17]. IR-induced DSBs are repaired mainly either by NHEJ or HR [
17]. NHEJ directly re-ligates the free-ends of the broken DNA without the need for a homologous template and, thus, it is an error-prone process [
43]. To begin, NHEJ first requires the recruiting of the Ku70/Ku80 heterodimer to each end of the broken DNA and the formed complex triggers subsequent recruiting of DNA-PKcs that results in the juxtaposition of the two DNA ends. The Ku70/Ku80/DNA-PKcs complex further recruits the DNA ligase complex (XRCC4/XLF/DNA ligase IV/PNK) to process the final ligation [
43]. In contrast to NHEJ, HR takes advantage of sequence information present in the intact sister chromatid, accurately repairing DSBs with high fidelity [
43]. Thus, since NHEJ does not require a DNA template for the repair, it can function through the cell cycle. In contrast, HR mainly operates during the S and G2 phases when a DNA template becomes available after the DNA replication [
43]. Radiation also produces SSBs, which are mainly caused by base oxidation by ROS/RNS [
19]. To repair this type of damage, the cell uses the base excision repairing mechanism. To process the repair, the single and multiple damaged bases will first be removed by DNA glycosylase-mediated incision and apurinic endonuclease 1 (APE1)-mediated incision, respectively, and the generated nicks will be filled up by the joint work of DNA polymerases and the DNA ligase [
44]. In the end, the successful repair of the damaged DNA caused by IR permits cells to survive and reenter the cell cycle. On contrary, failure of repairing the damaged DNA will result in one of the following outcomes: senescence, autophagy, necrosis, or apoptosis (
Figure 2).