Chronic alcohol consumption and alcohol-associated liver disease (ALD) represent a major public health problem worldwide. Only a minority of patients with an alcohol-use disorder (AUD) develop severe forms of liver disease (e.g., steatohepatitis and fibrosis) and finally progress to the more advanced stages of ALD, such as severe alcohol-associated hepatitis and decompensated cirrhosis. Emerging evidence suggests that gut barrier dysfunction is multifactorial, implicating microbiota changes, alterations in the intestinal epithelium, and immune dysfunction. This failing gut barrier ultimately allows microbial antigens, microbes, and metabolites to translocate to the liver and into systemic circulation. Subsequent activation of immune and inflammatory responses contributes to liver disease progression.
- intestinal barrier
- chronic alcohol consumption
- gut-liver axis
- intestinal immunity
- inflammation
1. Introduction
2. The Gut Barrier
2.1. Gut Microbiome
2.2. Intestinal Epithelium
2.3. Intestinal Immune System
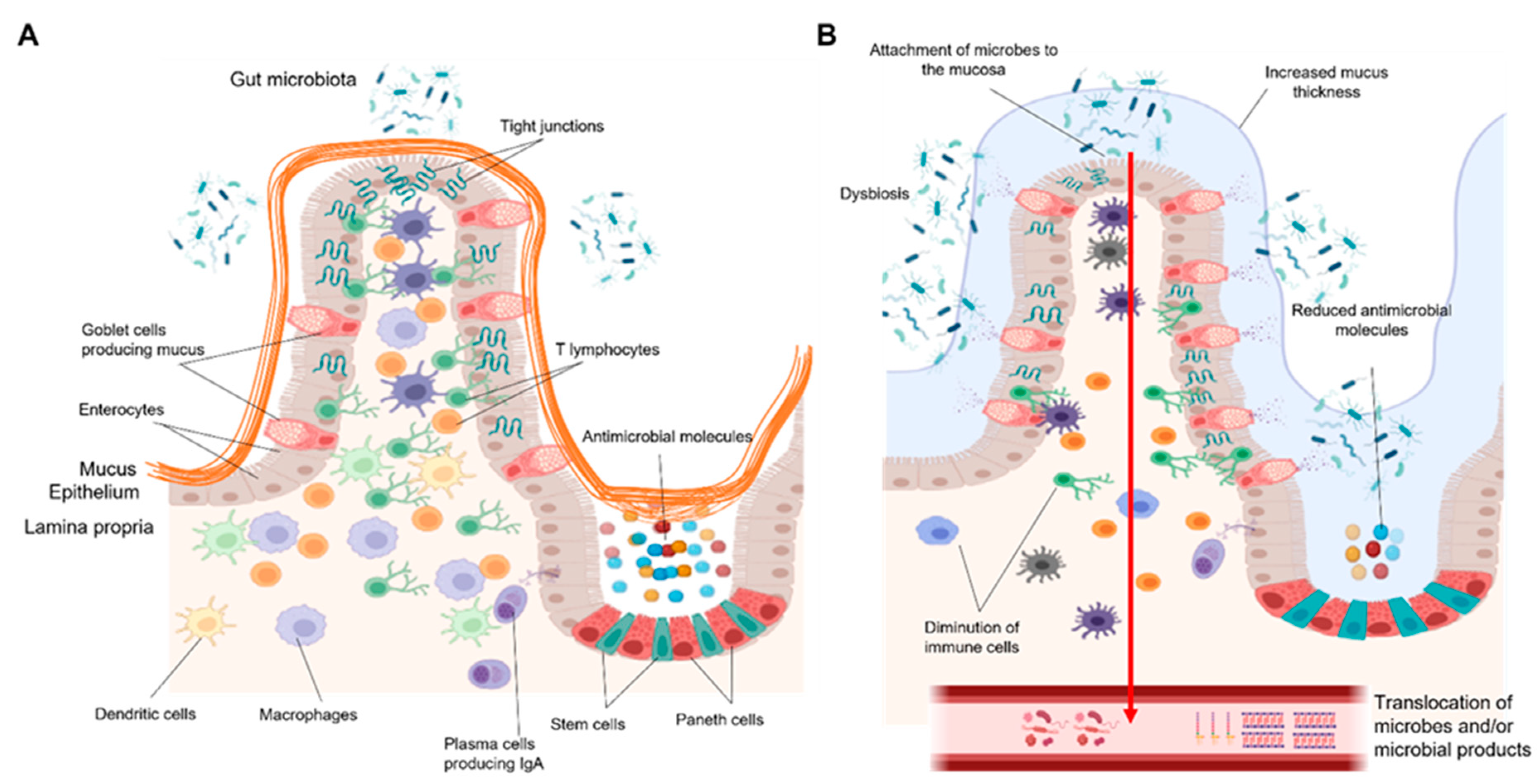
3. How Can Gut Barrier Dysfunction Contribute to ALD Progression?
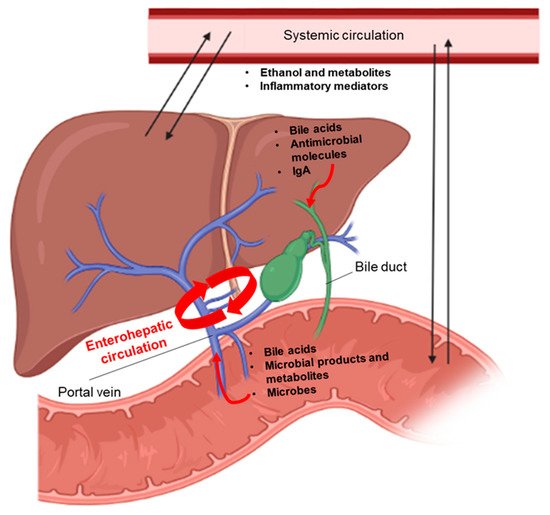
3.1. Systemic Inflammatory Responses
3.2. Impairment of Immune Responses Related to Microbes in the Liver
This entry is adapted from the peer-reviewed paper 10.3390/ijms222312687
References
- World Health Organization. Global Status Report on Alcohol and Health 2018; World Health Organization: Geneva, Switzerland, 2018.
- O’Shea, R.S.; Dasarathy, S.; McCullough, A.J.; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology 2010, 51, 307–328.
- Åberg, F.M.F.; Färkkilä, M. Drinking and Obesity: Alcoholic Liver Disease/Nonalcoholic Fatty Liver Disease Interactions. Semin. Liver Dis. 2020, 4, 154–162.
- Traversy, G.; Chaput, J.-P. Alcohol Consumption and Obesity: An Update. Curr. Obes. Rep. 2015, 4, 122–130.
- Singh, A.; Amin, H.; Garg, R.; Gupta, M.; Lopez, R.; Alkhouri, N.; McCullough, A. Increased Prevalence of Obesity and Metabolic Syndrome in Patients with Alcoholic Fatty Liver Disease. Dig. Dis. Sci. 2020, 65, 3341–3349.
- Peeraphatdit, T.B.; Ahn, J.C.; Choi, D.H.; Allen, A.M.; Simonetto, D.A.; Kamath, P.S.; Shah, V.H. A Cohort Study Examining the Interaction of Alcohol Consumption and Obesity in Hepatic Steatosis and Mortality. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2020; Volume 95, pp. 2612–2620.
- Hwang, S.; Ren, T.; Gao, B. Obesity and binge alcohol intake are deadly combination to induce steatohepatitis: A model of high-fat diet and binge ethanol intake. Clin. Mol. Hepatol. 2020, 26, 586–594.
- Lang, S.; Schnabl, B. Microbiota and Fatty Liver Disease—The Known, the Unknown, and the Future. Cell Host Microbe 2020, 28, 233–244.
- Jiang, L.; Stärkel, P.; Fan, J.-G.; Fouts, D.E.; Bacher, P.; Schnabl, B. The gut mycobiome: A novel player in chronic liver diseases. J. Gastroenterol. 2021, 56, 1–11.
- Yang, A.-M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841.
- Jiang, L.; Lang, S.; Duan, Y.; Zhang, X.; Gao, B.; Chopyk, J.; Schwanemann, L.K.; Ventura-Cots, M.; Bataller, R.; Bosques-Padilla, F.; et al. Intestinal Virome in Patients With Alcoholic Hepatitis. Hepatology 2020, 72, 2182–2196.
- Zhou, Z.; Zhong, W. Targeting the gut barrier for the treatment of alcoholic liver disease. Liver Res. 2017, 1, 197–207.
- Khoshbin, K.; Camilleri, M. Effects of dietary components on intestinal permeability in health and disease. Am. J. Physiol. Liver Physiol. 2020, 319, G589–G608.
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.; Jamar, F.; Stärkel, P.; Windey, K.; Tremaroli, V.; Bäckhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493.
- Maccioni, L.; Gao, B.; Leclercq, S.; Pirlot, B.; Horsmans, Y.; De Timary, P.; Leclercq, I.; Fouts, D.; Schnabl, B.; Stärkel, P. Intestinal permeability, microbial translocation, changes in duodenal and fecal microbiota, and their associations with alcoholic liver disease progression in humans. Gut Microbes 2020, 12, 1–23.
- Wilkin, R.J.; Lalor, P.F.; Parker, R.; Newsome, P.N. Murine Models of Acute Alcoholic Hepatitis and Their Relevance to Human Disease. Am. J. Pathol. 2016, 186, 748–760.
- Tabakoff, B.; Hoffman, P.L. Animal Models in Alcohol Research. Alcohol Res. Health J. Natl. Inst. Alcohol Abus. Alcohol. 2000, 24, 77–84.
- Cederbaum, I.A. Metabolism Alcohol. Clin. Liver Dis. 2013, 16, 667–685.
- Mestas, J.; Hughes, C.C.W. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738.
- Nguyen, T.L.A.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis. Model. Mech. 2015, 8, 1–16.
- Gao, B.; Xu, M.-J.; Bertola, A.; Wang, H.; Zhou, Z.; Liangpunsakul, S. Animal Models of Alcoholic Liver Disease: Pathogenesis and Clinical Relevance. Gene Expr. 2017, 17, 173–186.
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell 2012, 148, 1258–1270.
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340.
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591.
- Kamada, N.; Chen, G.Y.; Inohara, N.; Núñez, G. Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 2013, 14, 685–690.
- Fulde, M.; Hornef, M.W. Maturation of the enteric mucosal innate immune system during the postnatal period. Immunol. Rev. 2014, 260, 21–34.
- Ubeda, C.; Djukovic, A.; Isaac, S. Roles of the intestinal microbiota in pathogen protection. Clin. Transl. Immunol. 2017, 6, e128.
- Cotter, P.D.; Ross, R.; Hill, C. Bacteriocins—A viable alternative to antibiotics? Nat. Rev. Genet. 2013, 11, 95–105.
- Takiishi, T.; Fenero, C.M.; Câmara, N.O.S. Intestinal barrier and gut microbiota: Shaping our immune responses throughout life. Tissue Barriers 2017, 5, e1373208.
- Staley, C.; Weingarden, A.R.; Khoruts, A.; Sadowsky, M.J. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl. Microbiol. Biotechnol. 2017, 101, 47–64.
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168.
- Lin, R.; Liu, W.; Piao, M.; Zhu, H. A review of the relationship between the gut microbiota and amino acid metabolism. Amino Acids 2017, 49, 2083–2090.
- Allaire, J.; Crowley, S.M.; Law, H.T.; Chang, S.-Y.; Ko, H.-J.; Vallance, B.A. The Intestinal Epithelium: Central Coordinator of Mucosal Immunity. Trends Immunol. 2018, 39, 677–696.
- Gehart, H.; Clevers, H. Tales from the crypt: New insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 19–34.
- Hansson, G.C. Mucins and the Microbiome. Annu. Rev. Biochem. 2020, 89, 769–793.
- Belle, N.M.; Ji, Y.; Herbine, K.; Wei, Y.; Park, J.; Zullo, K.; Hung, L.-Y.; Srivatsa, S.; Young, T.; Oniskey, T.; et al. TFF3 interacts with LINGO2 to regulate EGFR activation for protection against colitis and gastrointestinal helminths. Nat. Commun. 2019, 10, 1–13.
- Herbert, D.R.; Yang, J.-Q.; Hogan, S.P.; Groschwitz, K.; Khodoun, M.; Munitz, A.; Orekov, T.; Perkins, C.; Wang, Q.; Brombacher, F.; et al. Intestinal epithelial cell secretion of RELM-β protects against gastrointestinal worm infection. J. Exp. Med. 2009, 206, 2947–2957.
- McDole, J.R.; Wheeler, L.W.; McDonald, K.G.; Wang, B.; Konjufca, V.; Knoop, K.; Newberry, R.D.; Miller, M.J. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nat. Cell Biol. 2012, 483, 345–349.
- Lu, R.; Zhang, Y.-G.; Xia, Y.; Zhang, J.; Kaser, A.; Blumberg, R.; Sun, J. Paneth Cell Alertness to Pathogens Maintained by Vitamin D Receptors. Gastroenterology 2020, 160, 1269–1283.
- George, J.J.; Oittinen, M.; Martin-Diaz, L.; Zapilko, V.; Iqbal, S.; Rintakangas, T.; Martins, F.T.A.; Niskanen, H.; Katajisto, P.; Kaikkonen, M.U.; et al. Polycomb Repressive Complex 2 Regulates Genes Necessary for Intestinal Microfold Cell (M Cell) Development. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 873–889.
- Wells, J.M.; Rossi, O.; Meijerink, M.; van Baarlen, P. Epithelial crosstalk at the microbiota-mucosal interface. Proc. Natl. Acad. Sci. USA 2011, 108, 4607–4614.
- Pelaseyed, T.; Hansson, G.C. Membrane mucins of the intestine at a glance. J. Cell Sci. 2020, 133, jcs240929.
- Bain, C.C.; Schridde, A. Origin, Differentiation, and Function of Intestinal Macrophages. Front. Immunol. 2018, 9, 2733.
- De Schepper, S.; Verheijden, S.; Aguilera-Lizarraga, J.; Viola, M.F.; Boesmans, W.; Stakenborg, N.; Voytyuk, I.; Schmidt, I.; Boeckx, B.; de Casterlé, I.D.; et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 2018, 175, 400–415.e13.
- Bekiaris, V.; Persson, E.K.; Agace, W.W. Intestinal dendritic cells in the regulation of mucosal immunity. Immunol. Rev. 2014, 260, 86–101.
- Hilpert, C.; Sitte, S.; Arnold, H.; Lehmann, C.H.K.; Dudziak, D.; Mattner, J.; Voehringer, D. Dendritic Cells Control Regulatory T Cell Function Required for Maintenance of Intestinal Tissue Homeostasis. J. Immunol. 2019, 203, 3068–3077.
- Aliberti, J. Immunity and Tolerance Induced by Intestinal Mucosal Dendritic Cells. Mediat. Inflamm. 2016, 2016, 1–8.
- Tezuka, H.; Ohteki, T. Regulation of IgA Production by Intestinal Dendritic Cells and Related Cells. Front. Immunol. 2019, 10, 1891.
- Paap, E.M.; Müller, T.M.; Sommer, K.; Neurath, M.F.; Zundler, S. Total Recall: Intestinal TRM Cells in Health and Disease. Front. Immunol. 2021, 11, 1–8.
- Stärkel, P.; Schnabl, B. Bidirectional Communication between Liver and Gut during Alcoholic Liver Disease. Semin. Liver Dis. 2016, 36, 331–339.
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut–liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411.
- Gao, B.; Tsukamoto, H. Inflammation in Alcoholic and Nonalcoholic Fatty Liver Disease: Friend or Foe? Gastroenterology 2016, 150, 1704–1709.
- Leclercq, S.; de Saeger, C.; Delzenne, N.; de Timary, P.; Stärkel, P. Role of inflammatory pathways, blood mononuclear cells, and gut-derived bacterial products in alcohol dependenc. Biol. Psychiatry 2014, 76, 725–733.
- Leclercq, S.; De Timary, P.; Delzenne, N.; Stärkel, P. The link between inflammation, bugs, the intestine and the brain in alcohol dependence. Transl. Psychiatry 2017, 7, e1048.
- Stärkel, P.; Schnabl, B.; Leclercq, S.; Komuta, M.; Bataller, R.; Argemi, J.; Palma, E.; Chokshi, S.; Hellerbrand, C.; Maccioni, L.; et al. Deficient IL-6/Stat3 Signaling, High TLR7, and Type I Interferons in Early Human Alcoholic Liver Disease: A Triad for Liver Damage and Fibrosis. Hepatol. Commun. 2019, 3, 867–882.
- Wang, M.; You, Q.; Lor, K.; Chen, F.; Gao, B.; Ju, C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J. Leukoc. Biol. 2014, 96, 657–665.
- Gao, B.; Ahmad, M.F.; Nagy, L.E.; Tsukamoto, H. Inflammatory pathways in alcoholic steatohepatitis. J. Hepatol. 2019, 70, 249–259.
- Altamirano, J.; Miquel, R.; Katoonizadeh, A.; Gonzalez-Abraldes, J.; Duarte–Rojo, A.; Louvet, A.; Augustin, S.; Mookerjee, R.; Michelena, J.; Smyrk, T.C.; et al. A Histologic Scoring System for Prognosis of Patients With Alcoholic Hepatitis. Gastroenterology 2014, 146, 1231–1239.e6.
- Stärkel, P.; Leclercq, S.; de Timary, P.; Schnabl, B. Intestinal dysbiosis and permeability: The yin and yang in alcohol dependence and alcoholic liver disease. Clin. Sci. 2018, 132, 199–212.
- Yang, L.; Eseki, E. Toll-Like Receptors in Liver Fibrosis: Cellular Crosstalk and Mechanisms. Front. Physiol. 2012, 3, 138.
- Takeda, K.; Akira, S. Toll-Like receptors. Curr. Protoc. Immunol. 2015, 2015, 14.12.1–14.12.10.
- Szabo, G. Gut–Liver Axis in Alcoholic Liver Disease. Gastroenterology 2015, 148, 30–36.
- Hritz, I.; Mandrekar, P.; Velayudham, A.; Catalano, D.; Dolganiuc, A.; Kodys, K.; Kurt-Jones, E.; Szabo, G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 2008, 48, 1224–1231.
- Inokuchi, S.; Tsukamoto, H.; Park, E.; Liu, Z.-X.; Brenner, D.A.; Seki, E. Toll-Like Receptor 4 Mediates Alcohol-Induced Steatohepatitis Through Bone Marrow-Derived and Endogenous Liver Cells in Mice. Alcohol. Clin. Exp. Res. 2011, 35, 1509–1518.
- Boetticher, N.C.; Peine, C.J.; Kwo, P.; Abrams, G.A.; Patel, T.; Aqel, B.; Boardman, L.; Gores, G.J.; Harmsen, W.S.; McClain, C.J.; et al. A Randomized, Double-Blinded, Placebo-Controlled Multicenter Trial of Etanercept in the Treatment of Alcoholic Hepatitis. Gastroenterology 2008, 135, 1953–1960.
- Gustot, T.; Lemmers, A.; Moreno, C.; Nagy, N.; Quertinmont, E.; Nicaise, C.; Franchimont, D.; Louis, H.; Devière, J.; Le Moine, O. Differential liver sensitization to Toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology 2006, 43, 989–1000.
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312.
- Ju, C.; Mandrekar, P. Macrophages and Alcohol-Related Liver Inflammation. Alcohol Res. Curr. Rev. 2015, 37, 251–262.
- Michelena, J.; Altamirano, J.; Abraldes, J.G.; Affò, S.; Morales-Ibanez, O.; Sancho-Bru, P.; Dominguez, M.; Garcia-Pagan, J.C.; Fernández, J.; Arroyo, V.; et al. Systemic inflammatory response and serum lipopolysaccharide levels predict multiple organ failure and death in alcoholic hepatitis. Hepatology 2015, 62, 762–772.
- Piano, S.; Singh, V.; Caraceni, P.; Maiwall, R.; Alessandria, C.; Fernandez, J.; Soares, E.C.; Kim, D.J.; Kim, S.E.; Marino, M.; et al. Epidemiology and Effects of Bacterial Infections in Patients With Cirrhosis Worldwide. Gastroenterology 2019, 156, 1368–1380.e10.