Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Health Care Sciences & Services
Hypertension is well known to alter the structure and function of cerebral blood vessels not only through its haemodynamics effects but also for its relationships with endothelial dysfunction, oxidative stress and inflammation.
- hypertension
- cerebral complications
- endothelial dysfunction
- oxidative stress
- neuroinflammation
- innate immune system
- Toll-like receptors
1. Pathogenetic Mechanisms of Hypertension–Brain Induced Complications: Traditional Mechanisms
1.1. Cerebral Blood Flow (CBF) Autoregulation
Regulating CBF in a normal brain ensures relatively constancy over a wide range of blood pressure changes [1]. Autoregulation is impaired when blood pressure exceeds the compensatory vasoconstrictor or vasodilatory capacity. This event usually occurs when the average blood pressure falls below 50 mm Hg or rises above 150 mm Hg in a normotensive subject (Figure 1).
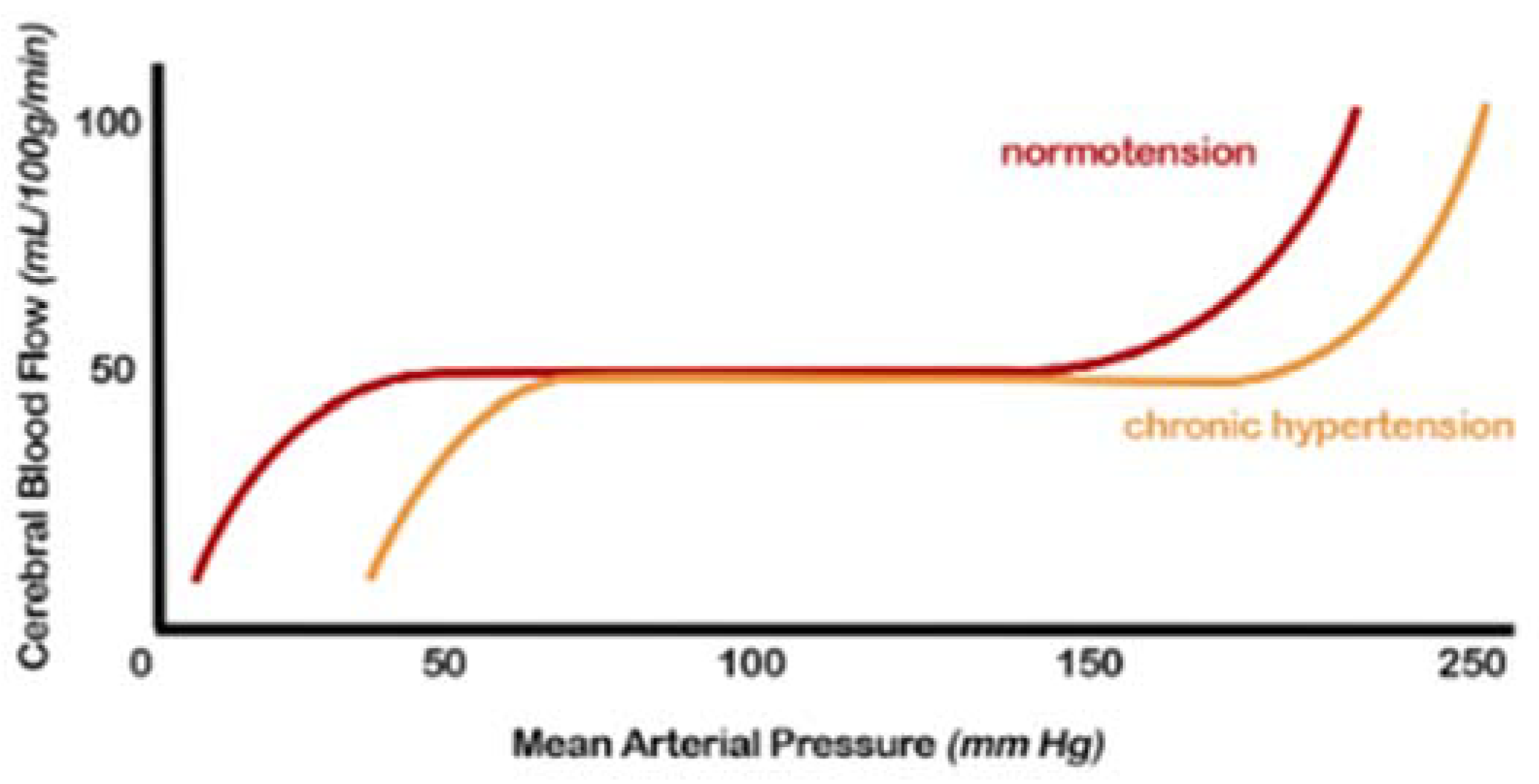
Figure 1. Cerebral autoregulation curve: in patients with hypertension, this range is “right-shifted”, or in other words, the normal range mean arterial pressure in which cerebral blood flow remains constant due to cerebral autoregulation is higher. Both the lower and upper limits are shifted (from [2], modified).
Furthermore, in the presence of hemodynamic compromise, vasodilation of the intracranial arterial vessels is observed. This underlying vasodilation limits the brain’s ability to further vasodilate in response to other ischemic events, resulting in an increased risk of subsequent accidents. Hypertensive patients show an altered cerebrovascular reserve, which can be corrected by appropriate antihypertensive therapy [1][3].
The exact mechanisms by which hypertension affects autoregulation of CBF are not fully understood but likely include alterations in myogenic tone and remodelling of internal vessels, increasing the wall-to-lumen ratio [2][4][5][1][3].
The cerebral autoregulatory response occurs primarily in the small vessels and has both myogenic and neurogenic components. Myogenic autoregulation occurs in the smallest arterioles and appears to be primarily responsible for cerebral autoregulation at blood pressure above the normal physiological range. On the other hand, neurogenic autoregulation acts at the level of large arterioles and small arteries by the autonomic perivascular sympathetic nerves. Endothelial dysfunction also causes excess nitric oxide (NO) production via upregulation of NO synthase. NO induces vasodilation, particularly in conditions of high intravascular flows, which would tend to counteract the protective effects of vasoconstriction. Some studies have also shown that NO can increase the permeability of cerebral vessels through a mechanism controlled by cyclic guanosine monophosphate (cGMP) [3][6].
1.2. Endothelial Dysfunction and Oxidative Stress
The main pathways involved in the main endothelial functions are reported in Table 1, and they include regulation of vascular tone, fibrinolysis and coagulation, inflammation and formation, repair and remodelling of blood vessels.
Table 1. Molecules produced and secreted by endothelial cells.
| Regulation of Vascular Tone |
|
|
|
| Balanced Blood Fluidity/Thrombosis |
|
|
|
| Vascular Inflammation and Immunological Process Control |
|
|
|
|
These endothelium functions, even if they are described separately, are closely interrelated.
The term endothelial dysfunction identifies the transition from a normal to a damaged endothelium that can express itself with a proinflammatory, constricting, proliferative and procoagulative phenotype. Endothelial dysfunction has been studied extensively in peripheral arteries and was found to precede the elevation in blood pressure and, once developed, to correlate with its severity and target organ damage, including brain damage [7][8][9][10][11][12].
The main factors underlying endothelial dysfunction are therefore represented by reduced bioavailability of NO, impaired smooth muscle response to vasodilators, increased production of vasoconstrictive substances or high shear stress [7][8][13][14][9][11]. As previously reported in the introduction section, the endothelium dysfunction is accompanied by increased production of NO supported by alterations in NO metabolism (high NO degradation, NO inactivation or the presence of NO inhibitors) and with a consequent increase in oxidative stress. Oxidative stress is a condition in which ROS production exceeds the capacity of the antioxidant defence system. Excessive ROS generation, reduced antioxidant capacity or a combination of both can lead to oxidative stress. Persistent oxidative stress can deplete the effectiveness of antioxidant molecules, inactivate the enzymes responsible for antioxidant action and, therefore, compromise the cellular defence system [12][15].
1.3. Mitochondrial Dysfunction
This mechanism plays an essential role in the occurrence of cerebrovascular damage induced by hypertension. The alterations in energy production that occur in brain cells represent the basis for several brain alterations. Brain mitochondria produce about 90% of the energy used by brain cells [16], necessary for the performance of their functions, such as intercellular communication and the transmission of stimuli and signals. An adequate energy supply by the mitochondria is essential for neuronal excitability and survival, so they are implicated in the pathogenesis of neurodegenerative diseases and cerebral ischemia.
Recent data have suggested a close link between excessive ROS generation and the development of neuronal death [17]. The brain is particularly prone to oxidative damage, both due to the lack of antioxidant defences [17] and because it can detect targets of free radicals derived from oxygen, such as the high traffic of calcium through neuronal membranes, presence of excitotoxic amino acids and auto-oxidizable neurotransmitters and a high quantity of polyunsaturated fatty acids contained within membrane lipids.
ROS contribute not only to the damage of macromolecules but also to the transduction of apoptotic signals. Overproduction of ROS by the respiratory chain of brain mitochondria can progressively impair mitochondrial energy metabolism in hypertension. This phenomenon may be implicated in the vulnerability to cerebral ischemia, resulting in progressive neuronal cell death [16].
The experimental evidence linking mitochondrial dysfunction with cerebral vascular damage in hypertensive is limited. However, the evidence that an assembly defect in mitochondrial complexes I and V has been demonstrated in the brain mitochondria of SHRs seems interesting [18]. To better understand the consequences of these data, it is necessary to consider the functions and the energetic cellular role of mitochondrial complexes I and V.
Mitochondrial complex I, or nicotinamide adenine dinucleotide phosphate (NADPH) ubiquinone oxidoreductase, is the most important of the mitochondria mediate oxidative phosphorylation (OXPHOS), contributing about 40% to the energy required for the synthesis of adenosine triphosphate (ATP) by ATP synthase [19]. On the other hand, mitochondrial complex V is responsible for the catalytic phosphorylation of adenosine diphosphate (ADP) to form ATP. Therefore, as a consequence of the assembly defects reported, a reduction in the production of ATP is present in the brains of the SHRs, which induces cellular energy deficiency [18].
It is also known that mitochondria contribute to the intracellular homeostasis of the calcium ion [20]. Some studies indicate that, in elderly SHRs, it is possible to document a reduction in the potential of the mitochondrial inner membrane and consequent impairment of the activity of calcium-dependent enzymes, such as mitochondrial isoform of nitric oxide synthase (mtNOS). These alterations can undoubtedly contribute to a progression of mitochondrial dysfunction and neuronal cell death during hypertension [20]. It is interesting to note that mtNOS, located within the mitochondrial membrane, is involved in cell apoptosis by modulating the transmembrane potential, inhibiting chaim respiration and ATP synthesis, and is correlated with the onset of brain damage induced by hypertension [21].
1.4. Microcirculation
In the course of arterial hypertension, it is possible to document alterations in the cerebral microcirculation consisting of functional alterations that influence the vasomotor capacity, usually capable of transiently modifying the blood flow, which sometimes leads to thrombotic episodes [22][2][23][4][24][25][3][26].
Critical deficits in global or regional brain perfusion induce suppression of brain activity and cognitive dysfunction [27]. Inflammation can also damage the vessel wall through alterations in endothelial function that result in a reduced function of the cerebral microcirculation with irreversible neuronal damage [28]. Cerebral microbleeds associated with hypertension are typically localized in the basal ganglia, thalamus, brainstem and cerebellum, while a lobar distribution linked to cerebral amyloid angiopathy is common [29]. Functional impairment of the endothelium appears to be an early indicator of vascular dysfunction in cSVD( cerebral small vessel disease) and large cerebral vessels [9][11][30]. Unfortunately, direct assessment of cerebral endothelial function in humans is not feasible, limiting the number of studies available. A recent study analysed cerebral and peripheral vessel reactivity measurements in response to CO2 inhalation using transcranial doppler and duplex ultrasound in lacunar stroke patients and control subjects. Abnormalities in peripheral artery reactivity appeared to be related to vascular risk factors, and the severity of endothelial dysfunction in the cerebral arteries was related to the onset of lacunar stroke in cSVD patients [31]. It has also been shown that endothelial function, assessed by NO metabolism analysis, is impaired in patients with lacunar infarction and associated cSVD [32].
As previously reported, ROS are key mediators of cerebrovascular dysfunction in hypertension, as they contribute to vascular rarefaction and structural remodelling of cerebral blood vessels and, therefore, lead to functional alterations with profound consequences for CBF. In particular, experimental studies, including models of hypertension in rodents, report that targeting the enzyme that produces ROS, NADPH oxidase or its assembly protects against cerebrovascular oxidative stress and, consequently, from alterations endothelium-dependent relaxation and functional hyperaemia [33][34]. However, existing studies showing the direct effect of hypertension on cerebral arterial tone [35] mainly describe endothelial dysfunction in isolated arteries; only a few studies show a direct link between endothelium-dependent vasodilation and CBF. In addition, postmortem histological findings, conducted in patients with severe cSVD, show in these subjects an intact endothelial layer in the small arteries and the frontal white matter, while an apparent loss of myocytes and other wall cells was documented [36].
These conflicting aspects require further studies in order to document the role of endothelial dysfunction of small cerebral arteries exposed to high blood pressure before and during overt cSVD.
1.5. Endothelial Activation and BBB(brain–blood barrier) Involvement
Regarding the role of BBB in cSVD, it is necessary to consider the different types of associated brain lesions and that the components mentioned above are closely related to each other. Presumably, endothelial dysfunction is one of the main determinants of cerebral vessels’ structural and functional alterations. As it was reported earlier, the imbalance between the production of vasodilator and vasoconstrictor molecules as well as the haemodynamic effects induced by hypertension lead to endothelial activation, a prerequisite for thrombo-inflammation and an early indicator of impaired BBB, which is usually accompanied by an increase in circulating adhesion molecules which condition the rolling, adhesion and migration of leukocytes [37]. Peripheral markers of endothelial activation include soluble vascular cell adhesion molecules-1 (sVCAM-1), soluble intercellular adhesion molecule-1 (sICAM-1), sP-selectin and sE-selectin, which are associated with cSVD markers and a reduction in cognitive performance in hypertensive patients. This fact indicates an essential role of endothelial activation in the pathogenesis of cSVD mediated by hypertension [38].
Levels of sICAM-1 appear to be an important marker for lacunar stroke and early neurological deterioration [32], even if its origin is not exclusively endothelial, which limits its specificity. More specific endothelial activation markers have emerged recently, and among them, sE-selectin seems to correlate with the number of microbleeds in patients with cSVD [39]. Other studies have shown an increase in adhesion molecules, the rolling of leukocytes along the cerebral vessels and the infiltration and accumulation of T cells in the perivascular spaces in hypertensive individuals, suggesting a causal link between endothelial activation and cerebrovascular dysfunction [40][41].
Other data indicate the presence of a thrombo-inflammatory state in elderly hypertensive subjects with cSVD. Thrombo-inflammation favours vascular occlusions, resulting in greater permeability of the BBB and triggering damage to the vessel wall with consequent vascular ruptures [42]. It has also been described that impairment of BBB resulting from alterations of its cellular components occurs during neurodegenerative diseases [43] but has also been associated with cSVD and lacunar infarcts [44]. However, it still remains uncertain whether BBB dysfunction is the main cause of the changes occurring in the cerebral microcirculation, since, to date, studies have only reported an increase in BBB permeability in cSVD associated with arterial hypertension [45][46][47]. Nevertheless, the simultaneous presence in hypertensive subjects of an increased number of activated circulating immunocompetent cells, of endothelial activation and the presence of microglial activation in the central nervous system indicates the existence of a causal relationship between elevated arterial pressure and impaired BBB [22][2][5][48].
2. Pathogenetic Mechanisms of Hypertension—Brain Induced Complications: New Factors
2.1. Role of Neuroinflammation
The inflammatory response to stroke generally develops as a consequence of two sequential but closely related phenomena:
-
The activation of microglia and perivascular and parenchymal resident macrophages;
-
The infiltration of peripheral inflammatory cells into the brain.
Once activated, the glial cells can produce a large variety of proinflammatory mediators suitable for damaging the cerebral endothelium, which, in addition to constituting the BBB, is involved in the determinism and the modulation of the cerebral inflammatory response [47][49].
For example, releasing proinflammatory vasoactive mediators by the cerebral endothelium during the acute phase of stroke can induce blood clotting in the microcirculation, extending the infarct area. Furthermore, once the “neuroinflammatory” response induced by ischemic or haemorrhagic stroke is triggered, it progresses for several days after the onset of symptoms, contributing to the pathogenesis of the later stages of brain damage and causing a worsening neurological prognosis. These considerations support new therapeutic strategies for stroke for which a deepening of knowledge of the neuroinflammatory process is needed [50][51]. A variety of mediators involved in all stages of neuroinflammation, especially in ischemic stroke, have recently been reported from its initial stage to resolution. They include tumour necrosis factor-alfa (TNF-α), interleukin (IL) 1α, IL-1β, chemokine (C-X-C motif) ligand 7 (CXCL7), chemokine (C-C motif), ligand 5 (CCL5), chemokine (C-X-C motif) ligand 4 (CXCL4), chemokine (C-X3-C motif) ligand 1 (CX3CL1), adhesion molecules, proteases, prostanoids and leukotrienes, while IL-1, IL-10, TNF-α, IL-6, IL-20, IL-17, NADPH oxidase, chemokine (C-X-C motif) ligand 8 (CXCL8), inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) are involved in the enhancement phase of this process. In addition, other molecules, including transforming growth factor β (TGF-β), IL-17, IL-10 and IL-23, mediate the resolution phase [52][53][54][55][56][57][58][59].
Finally, some molecules such as the neutrophil-to-lymphocyte ratio family pyrin domain-containing 3 (NLRP3) inflammasome, Dickkopf WNT Signalling Pathway Inhibitor 3 (DKK-3), dectin-1, MKEY and microRNAs (miRNAs), as well as being implicated in the course of neuroinflammation [52][53][50][54][55][56][57][58][59], have shown to have interesting characteristics that can attribute a target role in the management of brain damage, and they are summarized as follows:
-
The NLRP3 inflammasome is responsible for the activation of IL-1β and the release of IL-18, which phosphorylate insulin receptor substrate 1 (IRS-1), worsening insulin resistance and causing neuronal death. The NLRP3 inflammasome is one of the primary mediators contributing to the neuroinflammation process and consequent brain damage [54].
-
DKK-3 concentrations are associated with endothelial dysfunction and atherosclerosis. High or low levels of DKK-3 are able of inducing a worsening of outcomes after ischemic stroke [55].
-
The interaction between Dectin-1 and damage-associated molecular patterns (DAMPs) determines the phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) and, subsequently, of spleen tyrosine kinase (SYK9), a kinase able to mediate the neuroinflammatory cascade through the release of some cytokines. Therefore, the inflammatory pathway mediated by Dectin-1/SYK plays a fundamental role in postictal neuroinflammation [56].
-
The heterodimer CXCL4-CCL5 plays a crucial role in developing brain damage [57].
-
MKEY, a cyclic peptide synthesized in mice, can avoid the formation of the heterodimer CXCL4-CCL5, thereby limiting ischemic brain injury and improving neurological deficits [58].
-
The expression of some miRNAs, such as 126, 124-3p, 30a and 16, is considerably elevated in patients with acute ischemic brain injury, even if they still cannot be successfully used in clinical routine for obvious reasons, above all being the high cost and their execution in highly specialized laboratories [59].
2.2. Role of the Innate Immune System
Several examples have emerged to support the activation and contribution of TLRs (TLRs Toll-like receptors) to the progression of vascular atherothrombotic diseases. The vasculature system expresses all of the known human TLRs, and under vascular pathophysiology, the levels of TLRs are found to be upregulated (Figure 2).
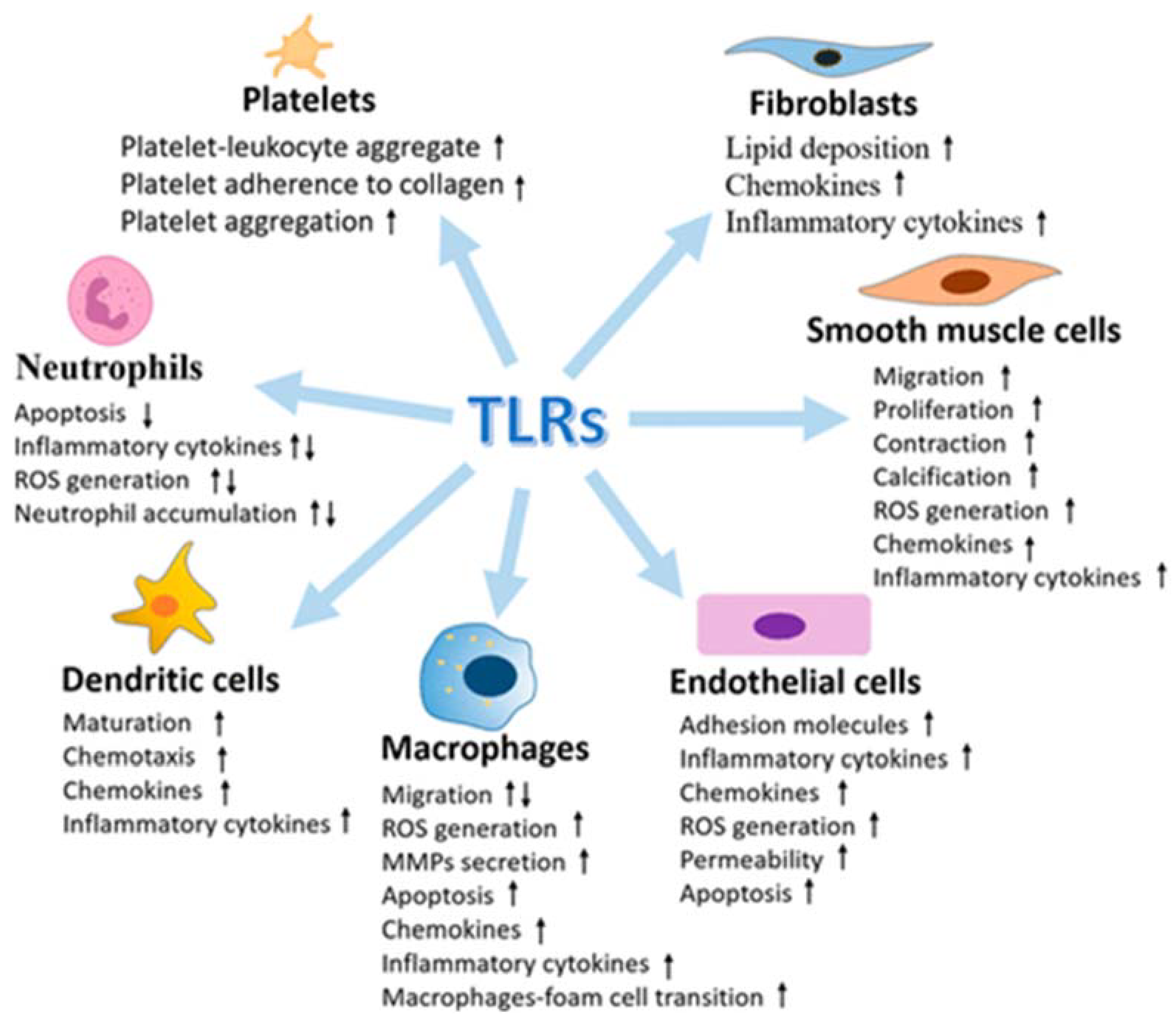
Figure 2. Multiple effects of Toll-like receptors (TLRs) in vasculature cells (from [60], modified).
2.2.1. TRLs: Discovery, Structure and Function
The Toll receptors are named for their structural similarity to Toll, a receptor first discovered in the Drosophila melanogaster, through a mutation in the Toll gene, caused abnormal development [61]. The embryos carrying the mutation were named “Toll”; later, a human homolog more closely related to Drosophila Toll was cloned [62], and the “human Toll” was then renamed “TLR4”.
TRLs, as previously reported, are responsible for recognizing and initiating an inflammatory response to microbial components expressed by bacteria, fungi, protozoa and viruses, as well as to DAMPs released by dying cells or generated as a result of tissue injury and oxidation [63]. In addition, the low complexity of the TLR signal, which includes four adapter molecules and three downstream inflammatory transcription factors, represents an efficient means of upregulating proinflammatory genes [64][60][63].
Inflammatory genes expressed as a result of TLRs activation include cytokines, whose expression patterns drive the adaptive immune response (cell-mediated Th1 response or humoral/antibody Th2 response), chemokines (chemotactic cytokines) that drive cell migration to target tissues and cell adhesion molecules that promote binding, rolling and infiltration of immune cells into the vascular wall and translocation to end organs [65].
TLRs are expressed on specialized immune cells (for example, macrophages and dendritic cells) and nonimmune cells (for example, epithelial, fibroblast and ECs).
TLRs are transmembrane proteins, or more specifically, type-I integral membrane glycoproteins with three structural domains:
- (1)
-
An amino (N)-terminal ectodomain that contains leucine-rich repeats and mediates ligand recognition;
- (2)
-
A single transmembrane domain that determines cellular localization;
- (3)
-
A carboxyl (C)-terminal cytoplasmic domain of the Toll/interleukin-1 receptor (TIR) that mediates downstream signalling.
So far, it has been shown that there are 10 TLR genes in humans (TLR1–TLR10) and 12 (TLR1–TLR9, TLR11–TLR13) in mice [66]. In general, TLRs can be divided into two groups based on their cellular location when detecting their respective ligands as follows:
Cell surface TLRs respond to microbial membrane materials such as lipids, lipoproteins and proteins, while intracellular TLRs recognize nucleic acids. Collectively, TLRs provide rigorous surveillance of intracellular and extracellular compartments, detecting most viral and bacterial molecular signatures as well as host-derived molecules released from damaged and apoptotic cells.
Most TLRs are homodimeric, although some of them can form heterodimers. Ligand binding promotes engagement of the two TIR domains of the cytosolic site of each receptor, and as the TIR domains get closer, a new signalling platform is created [68]. The formation of this platform is required for the recruitment of adapters containing cytosolic TIR domains, a key step in the TLRs signal. Several transmembrane proteins play the role of co-receptors in the TLRs signal, and the ability of TLRs to cooperate with accessory proteins increases the range of ligands that TLRs can recognize. Among them, particular importance is attributed to the myeloid differentiation primary response gene88-dependent pathway (MyD 88) and the Toll/interleukin1 receptor domain-containing adapter protein (TIRAP) [62][63][67].
The recruitment of adapter proteins represents the initial phase of TLRs signal transduction and, consequently, the first step in activating the innate immune system. Indeed, TLR4 activates the pathways dependent on MyD88 (myeloid differentiation primary response gene88-dependent pathway) and TIRAP, which lead to the production, respectively, of proinflammatory cytokines and type-I interferon. Various regulatory mechanisms are active in harmonizing the TLRs signal and avoiding an exaggerated immune response. Loss or deficiency of these regulatory mechanisms can be involved in developing immune-mediated and inflammatory diseases [64].
2.2.2. Mechanisms of DAMPs Presentation
DAMPs are endogenous molecules that are usually contained within cell membranes and protected from exposure to components of the immune system. However, when a cell is stressed or its plasma membrane is damaged, these endogenous molecules can be expressed on cell surfaces or diffuse freely into the extracellular space. The immune system recognizes these molecules as a danger and induces a response [69]. In most cases in hypertensive subjects, cell death represents the triggering mechanism of inflammation induced by the release of DAMPs with consequent brain damage. Circulating DAMPs released after hypoxia, trauma and cell death result in activation of TLRs in vascular smooth muscle, immune and endothelial cells (Figure 3).
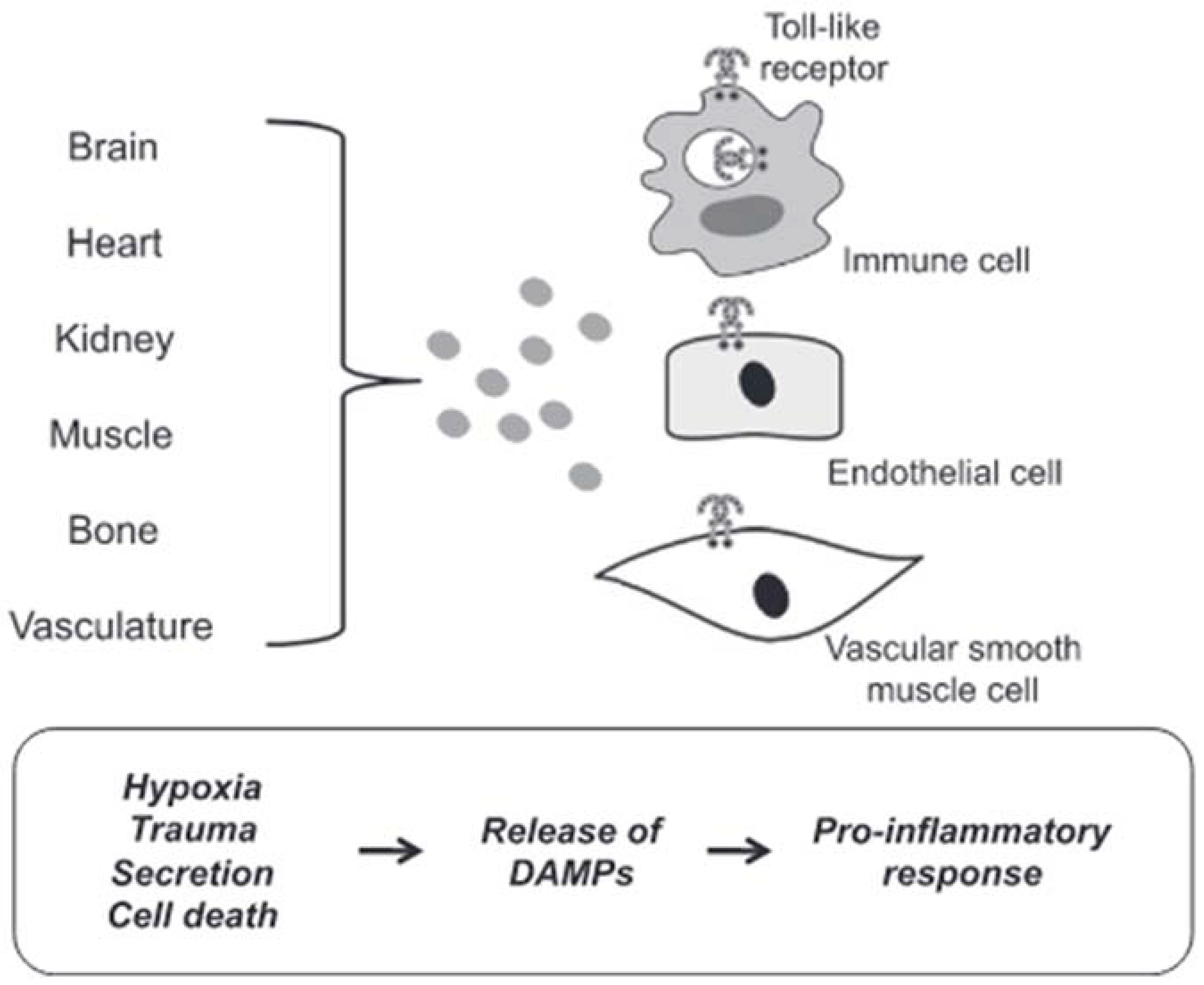
Figure 3. DAMPs induced activation of TLRs. Circulating DAMPs released after hypoxia, trauma and cell death lead to TLRs activation in immune cells, endothelial cells and vascular smooth muscle cells. Prolonged or excessive activation of TLRs on these cells provides a proinflammatory state, leading to endothelial dysfunction and subsequent cardiovascular disease (from [64], modified).
Prolonged or excessive activation of TLRs on these cells induces a proinflammatory state leading to endothelial dysfunction and subsequent cardiovascular disease [70]. Although there are many forms of cell death, necrotic cell death was generally thought to be the primary source of proinflammatory DAMPs due to the disintegration of the plasma membrane and the release of intracellular constituents [71]. Apoptosis can also be immunogenic due to the programmed release of immunostimulating molecules [72]. In this case, the release of DAMPs can be passive in the extracellular environment due to cell death or a damaged extracellular matrix, both active in the extracellular environment or on the surface of cells (i.e., neoantigens). Mechanisms of secretion and exposure are the results of cell stress. For these reasons, cell death would not seem to be the only precursor of the participation of DAMPs in the pathophysiology of cardiovascular diseases, in general, and of brain damage induced by arterial hypertension, in particular.
2.2.3. TLRs and Brain Damage-Related Hypertension
The development and maintenance of arterial hypertension depend on the contribution of the kidneys, the autonomic nervous system and the vascular system. Uncontrolled immune system activation and inflammation have been proposed as a unifying mechanism between these three organs and systems. Therefore, TLRs represent potential candidates for mediating this aberrant inflammation [70].
Overall, available data support the participation of TLRs in the aetiology of arterial hypertension and related organ damage, including brain damage. In this regard, it is known that cerebral ischemia causes an acute inflammatory reaction, which can exacerbate the brain damage caused by stroke [73]. The regulation of inflammation after an ictal event is multifactorial and includes vascular effects, distinct cellular responses, cell death and chemotaxis. Several cellular stipitis are involved in this process, including neurons, astrocytes, microglia and ECs, all of which respond to the resulting neuroinflammation in different ways [74]. TLRs are expressed on these brain cells and participate in the progression of brain damage through the following mechanisms:
This role of TLRs has been demonstrated by the results of some experimental studies, which can be summarized as follows:
-
Ischemia causes an increase in the expression of TLR2 in neurons (118) and microglia associated with the lesion [77];
-
The neurological damage and deficits caused by a stroke were significantly lower in TLR2-deficient mice compared to wild-type controls [76];
-
Although acute ischemic lesions (24 to 72 h) have been observed to be smaller in TLR2-deficient mice, the subsequent innate immune response has been reported to be more pronounced, causing progression of the ischemic injury [78].
In regard to endosomal TLRs, it is possible to summarize the data from the literature as follows:
-
TLR3 induces neuroprotection against ischemia through preconditioning [79];
-
The expression of TLR7 and TLR8 is associated with a negative outcome with increased inflammatory responses in patients with acute ischemic stroke [80];
-
TLR8 agonist induces increased neuronal cell death during oxygen or glucose deprivation, neurological deficit and T cell infiltration after stroke [81];
-
TLR9 activation induces neuroprotection against ischemic damage by increasing serum TNF-α by activating PI3K [84].
2.2.4. The Potential Therapeutic Role of TLRs in Cardiovascular Disorders
Molecular immunology and pathophysiology studies have provided new insights into the structural characteristics of TLRs, their ligands, their co-receptors and associated signalling proteins. TLR signalling triggers the transcriptional activation of different proinflammatory cytokines. Therefore, targeting the TLR signalling pathway is a plausible and complementary strategy to manage abnormal inflammation and vascular conditions. Therefore, both TLR agonists (inducers of protective immunity) and antagonists (suppressors of excessive inflammation) have been shown to have beneficial effects in various clinical conditions, such as cancer, microbial inflammation, autoimmunity and allergies, by modulating the tissue inflammatory response, while drug development targeting TLR regulation for cardiovascular disease is still in its infancy [64][60][62][63]. Nonetheless, new TLR inhibitory peptides have been identified capable of blocking the signalling of TLRs and suppressing the production of inflammatory cytokines [85], as well as mimetic peptides that seem to be able to increase stability, internalization and the sensitivity of the receptor, thus interrupting the interaction between TIR and MyD88 [86]. Furthermore, experimental data indicate that treatment with such peptides may protect against left ventricular dilation and hypertrophy in a mouse model of acute myocardial infarction [87].
Abbreviations
| ADP | adenosine diphosphate |
| ATP | adenosine triphosphate |
| BBB | brain–blood barrier |
| CBF | cerebral blood flow |
| CCL5 | chemokine (C-C motif) ligand 5 |
| cGMP | cyclic guanosine monophosphate |
| CRP | C-reactive protein |
| cSVD | cerebral small vessel disease |
| CX3CL1 | chemokine (C-X3-C motif) ligand 1 |
| CXCL4 | chemokine (C-X-C motif) ligand 4 |
| CXCL7 | chemokine (C-X-C motif) ligand 7 |
| CXCL8 | chemokine (C-X-C motif) ligand 8 |
| COX-2 | cyclooxygenase-2 |
| DAMPs | damage-associated molecular patterns |
| DKK-3 | Dickkopf WNT Signalling Pathway Inhibitor 3 |
| ECs | endothelial cells |
| ERF | endothelial releasing factors |
| IL | interleukine |
| iNOS | inducible nitric oxide synthase |
| IFN-α | interferone-α |
| IRS-1 | insulin receptor substrate 1 |
| ITAMs | immunoreceptor tyrosine-based activation motifs |
| miRNAs | microRNAs |
| MyD88 | myeloid differentiation primary response gene88-dependent pathway |
| mtDNA | mitochondrial DNA |
| mtNOS | mitochondrial isoform of nitric oxide synthase |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NLR | neutrophil-to-lymphocyte ratio |
| NLRP3 | NLR family pyrin domain-containing 3 |
| NO | nitric oxide |
| OXPHOS | mitochondria mediate oxidative phosphorylation |
| ROS | reactive oxygen species |
| SHRs | spontaneously hypertensive rats |
| SYK | spleen tyrosine kinase |
| TIR | Toll/interleukin-1 receptor |
| TIRAP | Toll-interleukin 1 receptor domain-containing adapter protein |
| TLRs | Toll-like receptors |
| TNF-α | tumour necrosis factor-alfa |
| TGF-β | transforming growth factor-beta |
| sICAM-1 | soluble intercellular adhesion molecule-1 |
| sVCAM-1 | soluble vascular cell adhesion molecules-1 |
| VSMCs | vascular smooth muscle cells |
This entry is adapted from the peer-reviewed paper 10.3390/ijms23052445
References
- Feldmann, E.; Skolnick, B.E. Cerebral hemodynamics, autoregulation, and blood pressure management. J. Stroke Cerebrovasc. Dis. 1999, 8, 176–182.
- Kelly, D.M.; Rothwell, P.M.M. Blood pressure and the brain: The neurology of hypertension. Pract. Neurol. 2020, 20, 100–111.
- Veglio, F.; Paglieri, C.; Rabbia, F.; Bisbocci, D.; Bergui, M.; Cerrato, P. Hypertension and cerebrovascular damage. Atherosclerosis 2009, 205, 331–341.
- Yu, J.G.; Zhou, R.R.; Cai, G.J. From Hypertension to Stroke: Mechanisms and potential prevention strategies. CNS Neurosci. Ther. 2011, 17, 577–584.
- Pistoia, F.; Sacco, S.; Degan, D.; Tiseo, C.; Ornello, R.; Carole, A. Hypertension and stroke: Epidemiological aspects and clinical evaluation. High Blood Press. Cardiovasc. Prev. 2016, 23, 9–18.
- Talman, W.T.; Dragon, D.N. Neuronal nitric oxide mediates cerebral vasodilatation during acute hypertension. Brain Res. 2007, 1139, 126–132.
- Vaziri, N.D.; Rodriguez-Iturbe, B. Mechanisms of disease: Oxidative stress and inflammation in the pathogenesis of hypertension. Nat. Clin. Pract. Nephrol. 2006, 2, 582–593.
- Colomba, D.; Duro, G.; Corrao, S.; Argano, C.; Di Chiara, T.; Nuzzo, D.; Pizzo, F.; Parrinello, G.; Scaglione, R.; Licata, G. Endothelial nitric oxide synthase gene polymorphisms and cardiovascular damage in hypertensive subjects: An Italian case-control study. Immun. Ageing 2008, 5, 4.
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. Adv. Exp. Med. Biol. 2017, 2, 511–540.
- Maida, C.D.; Vasto, S.; Di Raimondo, D.; Casuccio, A.; Vassallo, V.; Daidone, M.; Del Cuore, A.; Pacinella, G.; Cirrincione, A.; Simonetta, I.; et al. Inflammatory activation and endothelial dysfunction markers in patients with permanent atrial fibrillation: A cross-sectional study. Aging 2020, 12, 8423–8433.
- Poggesi, A.; Pasi, M.; Pescini, F.; Pantoni, L.; Inzitari, D. Circulating biologic markers of endothelial dysfunction in cerebral small vessel disease: A review. J. Cereb. Blood Flow Metab. 2016, 36, 72–94.
- Licata, G.; Di Chiara, T.; Licata, A.; Triolo, G.; Argano, C.; Parrinello, G.; Pinto, A.; Corrao, S.; Duro, G.; Scaglione, R. Relationship between circulating E-selectin, DD genotype of angiotensin converting enzyme, and cardiovascular damage in central obese subjects. Metabolism 2003, 52, 999–1004.
- Nadar, S.K.; Tayebjee, M.H.; Messerli, F.; Lip, G.Y. Target organ damage in hypertension: Pathophysiology and implications for drug theraphy. Curr. Pharm. Des. 2006, 12, 1581–1592.
- Tuttolomondo, A.; Daidone, M.; Pinto, A. Endothelial dysfunction and inflammation in ischemic stroke pathogenesis. Curr. Pharm. Des. 2020, 26, 4209–4219.
- González, J.; Valls, N.; Brito, R.; Rodrigo, R. Essential hypertension and oxidative stress: New insights. World J. Cardiol. 2014, 6, 353–366.
- Ohtsuki, T.; Matsumoto, M.; Suzuki, K.; Taniguchi, N.; Kamada, T. Mitochondrial lipid peroxidation and superoxide dismutase in rat hypertensive target organs. Am. J. Physiol. 1995, 268, H1418–H1421.
- Patten, D.A.; Germain, M.; Kelly, M.A.; Slack, R.S. Reactive oxygen species: Stuck in the middle of neurodegeneration. J. Alzheimers Dis. 2010, 20, S357–S367.
- Lopez-Campistrous, A.; Hao, L.; Xiang, W.; Ton, D.; Semchuk, P.; Sander, J.; Ellison, M.J.; Fernandez-Patron, C. Mitochondrial dysfunction in the hypertensive rat brain: Respiratory complexes exhibit assembly defects in hypertension. Hypertension 2008, 51, 412–419.
- Hunte, C.; Zickermann, V.; Brandt, U. Functional modules and structural basis of conformational coupling in mitochondrial complex I. Science 2010, 329, 448–451.
- Calderón-Cortés, E.; Cortés-Rojo, C.; Clemente-Guerrero, M.; Manzo-Avalos, S.; Villalobos-Molina, R.; Boldogh, I.; Saavedra-Molina, A. Changes in mitochondrial functionality and calcium uptake in hypertensive rats as a function of age. Mitochondrion 2008, 8, 262–272.
- Aguilera-Aguirre, L.; González-Hernández, J.C.; Pérez-Vázquez, V.; Ramírez, J.; Clemente-Guerrero, M.; Villalobos-Molina, R.; Saavedra-Molina, A. Role of intramitochondrial nitric oxide in rat heart and kidney during hypertension. Mitochondrion 2002, 1, 413–423.
- Meissner, A. Hypertension and the brain: A risk factor for more than heart disease. Cerebrovasc. Dis. 2016, 42, 255–262.
- Lawes, C.M.; Vander Hoorn, S.; Rodgers, A. Global burden of blood-pressure-related disease, 2001. Lancet 2008, 371, 1513–1518.
- Dahlof, B. Prevention of stroke in patients with hypertension. Am. J. Cardiol. 2007, 100, S17–S24.
- Scuteri, A. Brain injury as end-organ damage in hypertension. Lancet Neurol. 2012, 11, 1015–1017.
- Feihl, F.; Liaudet, L.; Waeber, B.; Levy, B.I. Hypertension: A disease of the microcirculation? Hypertension 2006, 48, 1012–1017.
- Marshall, R.S. Effects of altered cerebral hemodynamics on cognitive function. J. Alzheimers Dis. 2012, 32, 633–642.
- Tian, G.H.; Sun, K.; Huang, P.; Zhou, C.M.; Yao, H.J.; Huo, Z.J.; Hao, H.F.; Yang, L.; Pan, C.S.; He, K.; et al. Long-term stimulation with electroacupuncture at DU20 and ST36 rescues hippocampal neuron through attenuating cerebral blood flow in spontaneously hypertensive rats. Evid.-Based Complement. Altern. Med. 2013, 2013, 482947.
- Greenberg, S.M.; Vernooij, M.W.; Cordonnier, C.; Viswanathan, A.; Al-Shahi Salman, R.; Warach, S.; Launer, L.J.; Van Buchem, M.A.; Breteler, M.M. Microbleed Study Group: Cerebral micro bleeds: A guide to detection and interpretation. Lancet Neurol. 2009, 8, 165–174.
- Chrissobolis, S.; Miller, A.A.; Drummond, G.R.; Kemp-Harper, B.K.; Sobey, C.G. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front. Biosci. 2011, 16, 1733–1745.
- Deplanque, D.; Lavallee, P.C.; Labreuche, J.; Gongora-Rivera, F.; Jaramillo, A.; Brenner, D.; Abboud, H.; Klein, I.F.; Touboul, P.J.; Vicaut, E.; et al. Lacunar-BICHAT Investigators: Cerebral and extracerebral vasoreactivity in symptomatic lacunar stroke patients: A case-control study. Int. J. Stroke 2013, 8, 413–421.
- Markus, H.S.; Hunt, B.; Palmer, K.; Enzinger, C.; Schmidt, H.; Schmidt, R. Markers of endothelial and hemostatic activation and progression of cerebral white matter hyperintensities: Longitudinal results of the Austrian stroke prevention study. Stroke 2005, 36, 1410–1414.
- Kazama, K.; Anrather, J.; Zhou, P.; Girouard, H.; Frys, K.; Milner, T.A.; Iadecola, C. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ. Res. 2004, 95, 1019–1026.
- Didion, S.P.; Faraci, F.M. Angiotensin II produces superoxide-mediated impairment of endothelial function in cerebral arterioles. Stroke 2003, 34, 2038–2042.
- Pires, P.W.; Jackson, W.F.; Dorrance, A.M. Regulation of myogenic tone and structure of parenchymal arterioles by hypertension and the mineralocorticoid receptor. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H127–H136.
- Hainsworth, A.H.; Oommen, A.T.; Bridges, L.R. Endothelial cells and human cerebral small vessel disease. Brain Pathol. 2015, 25, 44–50.
- van Hooren, K.W.; Spijkers, L.J.; van Breevoort, D.; Fernandez-Borja, M.; Bierings, R.; van Buul, J.D.; Alewijnse, A.E.; Peters, S.L.; Voorberg, J. Sphingosine-1-phosphate receptor 3 mediates sphingosine-1-phosphate induced release of Weibel-Palade bodies from endothelial cells. PLoS ONE 2014, 9, e91346.
- Shoamanesh, A.; Preis, S.R.; Beiser, A.S.; Vasan, R.S.; Benjamin, E.J.; Kase, C.S.; Wolf, P.A.; DeCarli, C.; Romero, J.R.; Seshadri, S. Inflammatory biomarkers, cerebral microbleeds, and small vessel disease: Framingham heart study. Neurology 2015, 84, 825–832.
- Rouhl, R.P.; Damoiseaux, J.G.; Lodder, J.; Theunissen, R.O.; Knottnerus, I.L.; Staals, J.; Henskens, L.H.; Kroon, A.A.; de Leeuw, P.W.; Tervaert, J.W.; et al. Vascular inflammation in cerebral small vessel disease. Neurobiol. Aging 2012, 33, 1800–1806.
- Wei, Z.; Spizzo, I.; Diep, H.; Drummond, G.R.; Widdop, R.E.; Vinh, A. Differential phenotypes of tissue-infiltrating T cells during angiotensin II-induced hypertension in mice. PLoS ONE 2014, 9, e114895.
- Trott, D.W.; Harrison, D.G. The immune system in hypertension. Adv. Physiol. Educ. 2014, 38, 20–24.
- Nagai, M.; Hoshide, S.; Kario, K. Association of prothrombotic status with markers of cerebral small vessel disease in elderly hypertensive patients. Am. J. Hypertens. 2012, 25, 1088–1094.
- Marques, F.; Sousa, J.C.; Sousa, N.; Palha, J.A. Blood-brain-barriers in aging and in Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 38.
- Wardlaw, J.M.; Doubal, F.; Armitage, P.; Chappell, F.; Carpenter, T.; Muñoz Maniega, S.; Farrall, A.; Sudlow, C.; Dennis, M.; Dhillon, B. Lacunar stroke is associated with diffuse blood-brain barrier dysfunction. Ann. Neurol. 2009, 65, 194–202.
- Weiner, H.L.; Selkoe, D.J. Inflammation and therapeutic vaccination in CNS diseases. Nature 2002, 420, 879–884.
- Tuttolomondo, A.; Maida, C.; Pinto, A. Inflammation and inflammatory cell recruitment in acute cerebrovascular diseases. Curr. Immunol. Rev. 2015, 11, 24–32.
- Tuttolomondo, A.; Pecoraro, R.; Casuccio, A.; Di Raimondo, D.; Buttà, C.; Clemente, G.; Della Corte, V.; Guggino, G.; Arnao, V.; Maida, C.; et al. Peripheral frequency of CD4+ CD28− cells in acute ischemic stroke. Medicine 2015, 94, e813.
- Iadecola, C.; Gottesman, R.F. Neurovascular and cognitive dysfunction in hypertension: Epidemiology, pathobiology and treatment. Circ. Res. 2019, 124, 1025–1044.
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Casuccio, A.; Di Bona, D.; Aiello, A.; Accardi, G.; Arnao, V.; Clemente, G.; Corte, V.D.; et al. HLA and killer cell immunoglobulin-like receptor (KIRs) genotyping in patients with acute ischemic stroke. J. Neuroinflamm. 2019, 16, 88.
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Arnao, V.; Pinto, A.; Licata, G. Inflammation in ischemic stroke subtypes. Curr. Pharm. Des. 2012, 18, 4289–4310.
- Tuttolomondo, A.; Di Sciacca, R.; Di Raimondo, D.; Serio, A.; D’Aguanno, G.; La Placa, S.; Pecoraro, R.; Arnao, V.; Marino, L.; Monaco, S.; et al. Plasma levels of inflammatory and thrombotic/fibrinolytic markers in acute ischemic strokes: Relationship with TOAST subtype, outcome and infarct site. J. Neuroimmunol. 2009, 215, 84–89.
- Tuttolomondo, A.; Puleo, M.G.; Velardo, M.C.; Corpora, F.; Daidone, M.; Pinto, A. Molecular biology of atherosclerotic ischemic strokes. Int. J. Mol. Sci. 2020, 21, 9372.
- Maida, C.M.; Norrito, R.L.; Daidone, M.; Tuttolomondo, A.; Pinto, A. Neuroinflammatory mechanisms in ischemic stroke: Focus on cardioembolic stroke, background, and therapeutic approaches. Int. J. Mol. Sci. 2020, 21, 6454.
- Pétrilli, V.; Dostert, C.; Muruve, D.A.; Tschopp, J. The inflammasome: A danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 2007, 19, 615–622.
- Monaghan, A.; Kioschis, P.; Wu, W.; Zuniga, A.; Bock, D.; Poustka, A.; Delius, H.; Niehrs, C. Dickkopf genes are coordinately expressed in mesodermal lineages. Mech. Dev. 1999, 87, 45–56.
- Ye, X.; Hao, Q.; Ma, W.-J.; Zhao, Q.-C.; Wang, W.-W.; Yin, H.-H.; Zhang, T.; Wang, M.; Zan, K.; Yang, X.-X.; et al. Dectin-1/Syk signaling triggers neuroinflammation after ischemic stroke in mice. J. Neuroinflamm. 2020, 17, 17.
- Koenen, R.R.; Von Hundelshausen, P.; Nesmelova, I.V.; Zernecke, A.; Liehn, E.A.; Sarabi, A.; Kramp, B.K.; Piccinini, A.M.; Paludan, S.R.; Kowalska, M.A.; et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat. Med. 2009, 15, 97–103.
- Terao, S.; Yilmaz, G.; Stokes, K.Y.; Russell, J.; Ishikawa, M.; Kawase, T.; Granger, D.N. Blood cell-derived RANTES mediates cerebral microvascular dysfunction, inflammation, and tissue injury after focal ischemia–reperfusion. Stroke 2008, 39, 2560–2570.
- Long, G.; Wang, F.; Li, H.; Yin, Z.; Sandip, C.; Lou, Y.; Wang, Y.; Chen, C.; Wang, D.W. Circulating miR-30a, miR-126 and let-7b as biomarker for ischemic stroke in humans. BMC Neurol. 2013, 13, 178.
- Zhou, Y.; Little, P.J.; Downey, L.; Afroz, R.; Wu, Y.; Ta, H.T.; Xu, S.; Kamato, D. The role of toll-like receptors in atherothrombotic cardiovascular disease. ACS Pharmacol. Transl. Sci. 2020, 3, 457–471.
- Anderson, K.V.; Jürgens, G.; Nüsslein-Volhard, C. Establishment of dorsal-ventral polarity in the Drosophila embryo: Genetic studies on the role of the Toll gene product. Cell 1985, 42, 779–789.
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397.
- Rifkin, I.R.; Leadbetter, E.A.; Busconi, L.; Viglianti, G.; Marshak-Rothstein, A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol. Rev. 2005, 204, 27–42.
- Goulopoulou, S.; McCarthy, C.G.; Webb, R.C. Toll-like receptors in the vascular system: Sensing the dangers within. Pharmacol. Rev. 2016, 68, 142–167.
- Lundberg, A.M.; Ketelhuth, D.F.; Johansson, M.E.; Gerdes, N.; Liu, S.; Yamamoto, M.; Akira, S.; Hansson, G.K. Toll-like receptor 3 and 4 signalling through the TRIF and TRAM adaptors in haematopoietic cells promotes atherosclerosis. Cardiovasc. Res. 2013, 99, 364–373.
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337.
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461.
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364.
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289.
- Lazaridis, A.; Gavriilaki, L.; Douma, S.; Gkaliagkousi, E. Toll-like receptors in the pathogenesis of essential hypertension. A forthcoming immune-driven theory in full effect. Int. J. Mol. Sci. 2021, 22, 3451.
- Miyake, Y.; Yamasaki, S. Sensing necrotic cells. Adv. Exp. Med. Biol. 2012, 738, 144–152.
- Davidovich, P.; Kearney, C.J.; Martin, S.J. Inflammatory outcomes of apoptosis, necrosis and necroptosis. Biol. Chem. 2014, 395, 1163–1171.
- Shichita, T.; Ito, M.; Yoshimura, A. Post-ischemic inflammation regulates neural damage and protection. Front. Cell Neurosci. 2014, 8, 319.
- Li, L.; Acioglu, C.; Heary, R.F.; Elkabes, S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav. Immun. 2021, 91, 740–755.
- Fadakar, K.; Dadkhahfar, S.; Esmaeili, A.; Rezaei, N. The role of Toll-like receptors (TLRs) in stroke. Rev. Neurosci. 2014, 25, 699–712.
- Tang, S.C.; Arumugam, T.V.; Xu, X.; Cheng, A.; Mughal, M.R.; Jo, D.G.; Lathia, J.D.; Siler, D.A.; Chigurupati, S.; Ouyang, X.; et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 13798–13803.
- Lehnardt, S.; Lehmann, S.; Kaul, D.; Tschimmel, K.; Hoffmann, O.; Cho, S.; Krueger, C.; Nitsch, R.; Meisel, A.; Weber, J.R. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J. Neuroimmunol. 2007, 190, 28–33.
- Bohacek, I.; Cordeau, P.; Lalancette-Hébert, M.; Gorup, D.; Weng, Y.C.; Gajovic, S.; Kriz, J. Toll-like receptor 2 deficiency leads to delayed exacerbation of ischemic injury. J. Neuroinflamm. 2012, 9, 191.
- Shi, H.; Gabarin, N.; Hickey, E.; Askalan, R. TLR-3 receptor activation protects the very immature brain from ischemic injury. J. Neuroinflamm. 2013, 10, 104.
- Brea, D.; Sobrino, T.; Rodríguez-Yáñez, M.; Ramos-Cabrer, P.; Agulla, J.; Rodríguez-González, R.; Campos, F.; Blanco, M.; Castillo, J. Toll-like receptors 7 and 8 expression is associated with poor outcome and greater inflammatory response in acute ischemic stroke. Clin. Immunol. 2011, 139, 193–198.
- Tang, S.C.; Yeh, S.J.; Li, Y.I.; Wang, Y.C.; Baik, S.H.; Santro, T.; Widiapradja, A.; Manzanero, S.; Sobey, C.G.; Jo, D.G.; et al. Evidence for a detrimental role of TLR8 in ischemic stroke. Exp. Neurol. 2013, 250, 341–347.
- Hyakkoku, K.; Hamanaka, J.; Tsuruma, K.; Shimazawa, M.; Tanaka, H.; Uematsu, S.; Akira, S.; Inagaki, N.; Nagai, H.; Hara, H. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 2010, 171, 258–267.
- Stevens, S.L.; Ciesielski, T.M.; Marsh, B.J.; Yang, T.; Homen, D.S.; Boule, J.L.; Lessov, N.S.; Simon, R.P.; Stenzel-Poore, M.P. Toll-like receptor 9: A new target of ischemic preconditioning in the brain. J. Cereb. Blood Flow Metab. 2008, 28, 1040–1047.
- Lu, C.; Ha, T.; Wang, X.; Liu, L.; Zhang, X.; Kimbrough, E.O.; Sha, Z.; Guan, M.; Schweitzer, J.; Kalbfleisch, J.; et al. The TLR9 ligand, CpG-ODN, induces protection against cerebral ischemia/reperfusion injury via activation of PI3K/Akt signaling. J. Am. Heart Assoc. 2014, 3, e000629.
- Avbelj, M.; Horvat, S.; Jerala, R. The role of intermediary domain of MyD88 in cell activation and therapeutic inhibition of TLRs. J. Immunol. 2011, 187, 2394–2404.
- Davis, C.N.; Mann, E.; Behrens, M.M.; Gaidarova, S.; Rebek, M.; Rebek, J., Jr.; Bartfai, T. MyD88-dependent and -independent signaling by IL-1 in neurons probed by bifunctional Toll/IL-1 receptor domain/BB-loop mimetics. Proc. Natl. Acad. Sci. USA 2006, 103, 2953–2958.
- Van Tassell, B.W.; Seropian, I.M.; Toldo, S.; Salloum, F.N.; Smithson, L.; Varma, A.; Hoke, N.N.; Gelwix, C.; Chau, V.; Abbate, A. Pharmacologic inhibition of myeloid differentiation factor 88 (MyD88) prevents left ventricular dilation and hypertrophy after experimental acute myocardial infarction in the mouse. J. Cardiovasc. Pharmacol. 2010, 55, 385–390.
This entry is offline, you can click here to edit this entry!