Ageing can be defined as a time-dependent decline in the functionality of the body. At the cellular level, its essence can be seen as a gradual loss of normal cell function accompanied by a series of ageing phenotypes. Breaking the dominance of the senescent microenvironment in the senescent organism and changing this balance to one dominated by the rejuvenating microenvironment has the opportunity to reprogram the metabolism of senescent cells and thus break the characteristic cycle of senescence within senescent cells.
- ageing
- senescence
- senolytics/senostatics
- SASP
1. Reprogramming-Based Therapies to Reverse Senescence
Partial reprogramming simultaneously lengthens telomeres, inhibits p53, and restores mitochondrial function [31]. Interestingly, the telomerase reverse transcriptase overexpression in transgenic mice (Sp53/Sp16/SArf/Tg Tert mice) showed improved tumour resistance and was found to prevent ageing-related degeneration (mainly atrophy) and inflammatory processes, higher blood levels of IGF1, and a reduction in γ-H2AX foci. Increased glucose tolerance and neuromuscular coordination cause a longer average lifespan [40]. The telomere–p53–PGC pathway and its downstream gene network regulate the functional state of multiple organs and ageing: increased levels of p53 (Trp53) lead to inhibition of peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1α). The germline deletion of p53 fully restores PGC network expression; PGC-1α expression restores mitochondrial respiration, cardiac function, and glucose allosterism [41]. Furthermore, reducing peroxisome proliferator-activated receptor-gamma coactivator-1beta (PGC-1b) attenuates cellular senescence-related phenotypes [4]. This implies that short-term cyclic expression of OSKM can rejuvenate senescent cells’ epigenome in vivo, reduce p16Ink4a and SASP, and affect various senescence-related regulatory pathways (such as mitochondria dysfunction, DNA damage, impaired protein folding, telomere shortening, and inflammation [31]), thus exerting a synergistic anti-ageing effect.
Due to the “asynchronous” character of ageing, senescent cells reprogramming preferentially affects the tissues that are first influenced by ageing (e.g., adipose tissue, the immune system, and fibroblasts [1,2]). We, therefore, start our discussion with adipose tissue (Figure 2). Ageing is often accompanied by a decline in subcutaneous adipocytes marked by the depletion of adipose precursor cells [42], which in turn causes a change in fat tissue distribution—i.e., more visceral white fat and less brown fat [43,44] as well as ectopic fat deposits [45]. This transformation leads to a vicious circle of producing an ageing microenvironment through an imbalance in the inflammatory state and cellular metabolic state associated with ageing and, consequently, a disruption of cellular homeostasis (proteostasis) [46].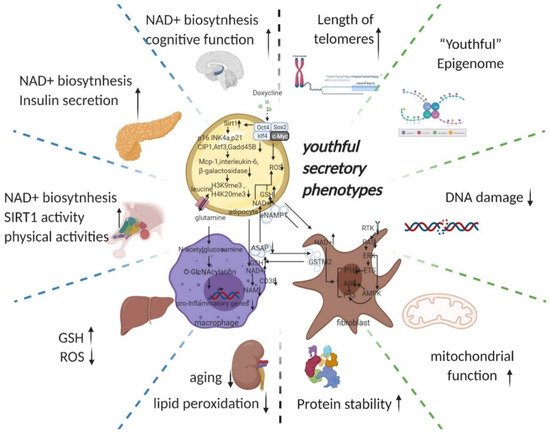
Figure 2. Potential intercellular mechanisms related to senescent cells specific reprogramming. Senescence in adipose precursor cells can be improved directly or indirectly (via reduced p21 and p16 pathways by overexpression of Sirt1 [43,55]) by doxycycline-induced overexpression of OSKM. The reversal of senescence by reprogramming can comprehensively improve senescence indicators (decrease in p16, p21, senescence-associated β-galactosidase, etc.) and can, at the same time, ameliorate senescence-associated secretory phenotypes (decreased Mcp-1 and Il-6, MMP13) and even improve histone methylation status (decrease in H3K9me3, H4K20me3) [31]. With the rejuvenation of adipose tissue (telomere lengthening, phenotypic rejuvenation remodelling, and promotion of gene damage repair), the upregulation of adipocyte glutaminase 1 [56] is reversed and the tissue is therefore rescued from the glutamine depleted state caused by ageing. Increased levels of glutamine will improve the chronic inflammatory state associated with ageing on a systemic scale by reducing the transcription of pro-inflammatory genes in macrophages in adipose tissue [48]. This means that the production of senescence-associated secretory phenotypes is reduced, thereby favouring the maintenance of a youthful state in surrounding fibroblasts, adipocytes, and themselves. The reprogramming also promotes the production of secretory eNAMPT in extracellular vesicles. By altering the NAD+ content of cells to regulate their mitochondrial metabolic state and redox homeostasis, eNAMPT promotes the rejuvenation of various cells throughout the body (improves pancreatic and hypothalamic secretion phenotypes, thereby amplifying anti-ageing effects via hormones) [49,57]. Macrophage rejuvenation not only improves the rejuvenation of the systemic secretory phenotype but also attenuates NAD+ degradation through reduced CD38 expression [11]. This may have a synergistic anti-senescence effect with eNAMPT. NAD+ and a rejuvenated secretory phenotype (possibly through metabolic reprogramming or cell rejuvenation via ERK–AMPK regulation of P16 and P53) improve the GST secretory capacity of fibroblasts. Delivery of GST to organs throughout the body via extracellular vesicles improved cellular redox homeostasis, resulting in a promising anti-ageing effect (improves liver redox status and kidney ageing) [51]. Taken together, local reprogramming through systemic cellular communication (eNAMPT, YSAP, and GST, etc.) produces synergistic anti-ageing effects (improvement in redox and metabolic imbalances caused by mitochondrial senescence and protein instability caused by ribosomal senescence). However, it is worth noting that further studies are needed to determine whether reprogramming can produce sufficient alterations in the secretory phenotype and whether intercellular communication can alter the secretory phenotype of adjacent cells. (black arrow: direct stimulatory, round arrow: cycle, dotted arrow: tentative stimulatory, down faded arrow: decrease, up faded arrow: increase; the grey dotted lines depict macro-level improvements on the left and micro-level improvements on the right, both separated by green dotted lines). (Created with BioRender.com)
Senescence of adipose precursor cells (caused by sirtuin 1 reduction) leads to the accumulation of senescent adipocytes [43], which secrete pro-inflammatory factors that constitute the first part of the senescent microenvironment and cause chronic inflammatory infiltration of adipose tissue [47]. As ageing redistributes fat (visceral fat increases), senescent adipose tissue carries the chronic inflammatory state associated with senescence (Mcp-1 and Il-6) throughout the body and gradually accumulates.
Increased white adipose tissue causes a decrease in glutamine levels in adipose tissue, leading to increased macrophage glycolysis in adipose tissue, increased pro-inflammatory transcription, and secretion of large amounts of SASP into the peripheral microcirculation, generating a second part of the senescent microenvironment [48].
M1 macrophages in senescent white adipose tissue consume large amounts of NAD+ [11,12], and adipocytes secrete less eNAMPT due to senescence [49,50], resulting in a systemic NAD+/NADH ratio imbalance (lower), which accelerates mitochondrial dysfunction-related senescence in cells throughout the body [3], resulting in an imbalance in energy metabolic status (glycolysis increase) and creating the third part of the senescent microenvironment.
Mitochondrial metabolic disorders cause enhanced glycolytic pathways and cellular redox disorders, resulting in systemic redox disorders [3,4]. Systemic fibroblasts under the influence of the first three parts of senescence and their own senescence, decrease GST secretion, exacerbating systemic peroxidation and creating the fourth part of the senescence microenvironment [51].
The redox disorder strongly affects genomic stability [5], generating many misconfigured proteins, which form aggregates that are expelled from the cells and adipocytes, which also discharge aged mitochondria, forming the fifth part of the senescent microenvironment [52,53,54].
Protein mismatches reach the upper limit of cellular discharge and continue to accumulate, damaging the cell’s genetic repair mechanism, which in turn disrupts all cellular functions into a state of irreversible death, releasing waste after its death and thus creating the sixth part of the senescent microenvironment. (Therefore, the simple removal of senescent cells might not avoid the senescence signal release process during senescent cell death).
2. Potential Key Mechanisms Related to Reprogramming-Based Therapies
2.1. Cyclin-Dependent Kinase Inhibitors (p16INK4A)
Multiple lines of evidence suggest p16INK4A extensive involvement in the ageing process, which could serve as an alternative regulatory centre in anti-ageing strategies. Mice with low levels of cell cycle checkpoint kinase BubR1 expression suffer from an acceleration in ageing as well as high levels of p16INK4A in tissues with age-related histopathology [58]. Targeted mutation of p16INK4A caused an ageing delay in the BubR1 mice. This delay went on together with reduced levels of senescent cells. This reveals a connection between biological ageing and cellular senescence [59]. In INK-ATTAC mice, where senescent cells expressing p16INK4A are specifically killed, the loss of senescent cells increases the lifespan and health span [60]. It has been observed that p16INK4A blocks E2F function and thus inhibits α-klotho promoter activity to accelerated senescence [61]. The p16INK4a prevents inactivation of retinoblastoma (Rb) phosphorylation by inhibiting cyclin D-dependent kinases. Then Rb represses E2F transcription factors expression by recruiting histone deacetylases to its promoter. The activated retinoblastoma (Rb) pathway simultaneously promotes formation of senescence-associated heterochromatic foci (SAHF), similarly refining the senescence-promoting mechanism of p16INK4a [62].
It is controversial whether there are side effects of p16INK4a -positive cells ablation. Removal of p16INK4a positive cells can lead to the side effect of fibrosis in the liver and perivascular tissue, which in turn reduces life expectancy [63]. Partial reprogramming of senescent cells is therefore one of the possible solutions to this problem.
2.2. Senescence-Associated Secretory Phenotype (SASP)
On the whole SASP is detrimental. For example, less than 1% of senescent preadipocytes can cause extensive physical dysfunction in young mice [13]. The killing of adjacent normal cells by SASP affects organ function [64], causing secondary senescence and increasing the accumulation of senescent cells that cause a variety of chronic inflammation/diseases [37] (senescent cells themselves are stalled in replication, so their primary cause of increased senescence is secondary senescence [64]). The anti-apoptotic capacity of senescent cells also increases the accumulation of senescent cells, and this property also protects these cells from SASP (creating a vicious circle) [65]. Targeted reprogramming of these cells may kill cells by breaking the anti-apoptotic capacity of senescent cells (it has been demonstrated in vivo in acute myeloid leukaemia cells, where short-term activation of OSKM expression induces apoptosis in leukaemic cells with little effect on normal haematopoietic stem and progenitor cells [66]). However, another possibility is to retain the beneficial components while eliminating the harmful ones (senescent cells are heterogeneous, and one subpopulation is beneficial for reprogramming and regeneration [14]). The validation of this hypothesis is one of the valuable directions for future research, so this section will comment on the beneficial potential of SASP.
Cells with p16INK4a promoter activation were monitored in vitro and in vivo to accumulate senescence and inflammation. They showed senescence features such as reduced cell proliferation and activation of senescence-associated β-galactosidase (SA-β-gal). Additionally, they augmented the expression of genes related to the SASP [67]. The biological conditions associated with ageing, p16Ink4a, create a relaxed tissue environment by producing the cytokine interleukin 6, which supports reprogramming of OSKM in vivo [6].
Skeletal muscle and (white) adipose tissue are two tissues that develop phenotypes associated with early senescence in response to BubR1 dysfunction, and they are high in p16INK4A and p19Arf [58]. p16INK4A inactivation in BubR1-deficient mice attenuated cellular senescence and premature senescence in these tissues. In contrast, p19Arf inactivation exacerbated senescence and senescence in BubR1 mutant mice. Thus, BubR1 functional incompetence triggers Cdkn2a locus activation in some mouse tissues [58]. p16INK4A/p19Arf-free tissue attenuates cellular senescence and reduces IL6 production and reprogramming efficiency. Tissues without p53, on the other hand, are extensively damaged and senescent, create high levels of IL6, and are efficiently reprogrammed. Thus p16INK4A, but not p19Arf, is required for OSKM-induced senescence and paracrine stimulation [68].
Specific removal of senescent cells, instead, reduces reprogramming effectiveness, and the outcome of senescence on reprogramming is mediated in part by interleukin-6 (IL-6) [7]. Thus, SASP promotes reprogramming, but reprogramming decreases SASP and thus can create a weak negative feedback regulation. On the plus side, it may prevent loss of organ function and teratomas caused by excessive reprogramming. However, it may also result in less efficient reprogramming. In addition to selecting safe tissues for reprogramming, one should also consider the extensive linkage of the selected tissue to the whole body and the simultaneous regulation of factors with synergistic effects in anti-ageing.
In addition to its contribution to reprogramming, SASP, as a major dynamic component of the senescence microenvironment, also assumes a role in regulating the cellular senescence state. Senescent cells can transfer proteins directly to neighbouring cells, and this cellular communication enhances the immune surveillance of cell senescence by natural killer (NK) cells [52]. A direct attempt to exploit this mechanism is to liberate NK cells from inhibition to target senescent cells for killing. This strategy shares a feature with two other strategies (i.e., first, targeting senescent cells for killing by means of chimeric antigen receptor T (CAR T) cells and NK cells [69], and second, 2-BCL-2 inhibition to induce apoptosis to kill senescent cells [70]).
Senescence transmission has been found to be transmitted via soluble factors and extracellular vesicles (sEVs) that makeup SASP [71]. In addition to SASP, Ras (rat sarcoma viral oncogene)-induced senescence through a juxtacrine NOTCH1 (Notch Receptor 1)–JAG1 (Jagged1) pathway contributes to senescence in adjacent cells, defined as “secondary senescence” [72,73]. This shows the intercellular transmission of the senescence state (NOTCH1/JAG1) and the potential of SASP secreted by senescent cells as a key node in anti-ageing strategies. After reprogramming deeply aged cells with OSKM, the senescent microenvironment is transformed into a rejuvenating microenvironment by altering SASP; multiple substances in the rejuvenating microenvironment metabolically remodel moderately aged cells (improved mitochondrial function) and then remodel mitochondrial nucleosome interactions such as ROS-DDR. The rejuvenation microenvironment is characterized by a wide range of substances that reshape the metabolism of moderately senescent cells (improved mitochondrial function). SASP is thus an essential part of the microenvironmental remodelling and intercellular communication; another part of SASP’s role is to link the endocrine (e.g., eNAMPT) and immune systems (e.g., glutamine) in this strategy to amplify the effects and scope of anti-ageing, which is described below (Figure 2).
2.3. DNA Methylation Level (Epigenetic Clock)
Epigenetics, characterized by acetylation and methylation (especially methylation of histone and the cytosines of CpG dinucleotides [35,74]), plays an essential role in cellular ageing. Thus, the “epigenetic clock (using the key age-related CpGs in a weighted linear model to predict chronological age)” might be indicative of biological age [37]. In addition, multiple studies have shown that epigenetic rejuvenation is possible through partial reprogramming, as reflected by age-deceleration in epigenetic clocks [37]. Therefore, epigenetic remodelling might be one of the most important ways to achieve a synergistic reversal of ageing. A genome-wide knockdown screen of human embryonic stem cells carrying a premature ageing mutation (CRISPR-Cas9-based) revealed that inactivation of the histone acetyltransferase KAT7 could inhibit p15INK4b transcription by reducing acetylation of histone H3 lysine 14 (H3K14) and is anti-ageing [75]. Reprogramming resets telomeres in supercentenarian cells, implying its massive role in cell rejuvenation [76]. Even with extensive epigenetic defects, reprogramming can still reset the epigenetic pattern to a revitalized pluripotent state [77].
2.4. Telomeres
In addition to remodelling the epigenetic landscape, partial reprogramming also prolongs telomeres in senescent cells [31]. Telomeres are repetitive nucleotide sequences located at the ends of chromosomes, are directly linked to cellular senescence, and are regulated by telomerase. Telomerase is a ribonucleoprotein complex that in humans consists of an enzyme, telomerase reverse transcriptase (TERT), plus a non-coding human telomerase RNA (hTR). The latter acts as a template for the prolonging of telomer length at the ends of chromosomes [78].
Tert overexpression significantly delayed ageing in mice by slowing telomere wear and preserving stem cell proliferative potential, but this required an increase in tumour suppression to counteract the pro-tumorigenic effects telomerase [40]. Abnormal telomere function inhibits PGC-1α and its downstream gene network via the p53-PGC pathway, thereby affecting cellular metabolism, causing organ dysfunction and leading to ageing [41]. Transient activation of telomerase restores neurogenesis in the subventricular zone and improves odour detection, suggesting that telomere lengthening reverses neural ageing and enhances its regenerative capacity, broadly improving organ function [79]. Therefore, tissue-specific transient telomere activation appears to be beneficial. For example, studies of telomerase gene therapy in mice by expressing pancreatic TERT with a broadly targeted adeno-associated virus (AAV) have also achieved beneficial effects, including increased pancreatic lifespan with reduced insulin sensitivity, osteoporosis, neuromuscular coordination, and molecular markers of ageing but no additional cancers occurred, which may suggest that the known oncogenic activity of telomerase is reduced when expressed in adult or aged organisms using an AAV vector [80].
In vitro assays with fibroblasts obtained from Tert knockout mice showed that mTert−/− cells are more susceptible to senescence and malignancy than mTert+/+ cells. Telomerase reverse transcriptase (TERT) expression is upregulated by mTert+/+ cells prior to senescence. In addition, knockdown or downregulation of TERT by CRISPR/Cas9 or shRNA reproduced the mTert−/− phenotype, while overexpression of TERT in mTert−/− cells was rescued [81]. In summary, whether transient induction of telomerase expression is beneficial should also be investigated in different tissues in vivo, but the vast differences in the oncogenic capacity of human and mouse limit the potential application of telomerase.
The activation of yes-associated protein 1 (YAP1), which upregulates the pro-inflammatory factor interleukin-18, can be rescued by mTert reactivation in mice with telomere dysfunction. In contrast, conventional SASP (IL-1, IL-6, IL8) did not show much change [82]. Thus, reprogramming strategies (which can regulate more inflammatory factors while lengthening telomeres) may have a synergistic effect on reducing the ageing-related chronic inflammation.
2.5. Youthful Secretory Phenotype (YSP)
The youthful secretory phenotype is a newly proposed hypothesis. It aims to generalize the anti-ageing factors (including GDF11, GPLD1, clusterin, Klotho, NAD+, eNAMPT, GSTM2, exosomes, et al.) found in young blood and in the secretome of young cells [20,21,22,23,49,51,57]. These “young factors (existing in young blood as well as being secreted by a heterogeneous subgroup of ageing cells)” may dilute or inhibit “old factors” promoting ageing, thus playing a rejuvenating role [14,15].
Plasma proteome alteration can also interfere with senescence through intercellular and organ–organ communication [19]. Exposure of aged mice to young serum improved regeneration of senescent satellite cells (through Notch signalling activation), increased senescent hepatocytes’ proliferation, and restored the cEBP-α complex to youthful levels [83]. It is shown that blood input from young donors to elderly recipients improves the latter’s senescence-related phenotype. It is not unique that the soluble factors and extracellular vesicles (sEVs) that make up SASP can influence other cells’ senescence state and even transmit senescence by secretion [71]. Therefore, it is tempting to think that the “youthful secretory phenotype (YSP)” (secreted by young cells) could also convey youth across the whole body. For example, neonatal umbilical cord (UC)-derived mesenchymal stem cell extracellular vesicles (MSC-EV), which are rich in anti-ageing rejuvenation signals, rejuvenate senescent adult bone marrow-derived mesenchymal stem cells (AB-MSC) [84]. Exposure of neonatal umbilical cord-derived MSC extracellular vesicles (UC-EV) increased telomere length in AB-MSC with a significant improvement in SASP. It improved age-related degeneration of mouse bones and kidneys at the organ level.
After blood alteration, cardiac hypertrophy and cardiomyocyte size decreased significantly in old mice with concomitant molecular remodelling [85]. Growth and differentiation factor 11 (GDF11) could be one of the “rejuvenating” factors in young blood. Restoring circulating GDF11 levels reverses functional and genetic damage in aged muscle stem cells [86]. However, it remains controversial whether blood exchange therapy works by restoring GDF11 in aged mice to youthful levels [86,87]. For example, whether young blood improves synaptic plasticity and cognitive function through the activation of cyclic AMP response element-binding protein (Creb) in dentate gyrus neurons [88] or by enhancing neurogenesis in ageing mice via GDF11 [20].
In addition, the mechanism of young blood combating senescence remains controversial. The autophagic activity of aged livers can be restored by exposure to plasma from juvenile donors. Conversely, inhibition of autophagic activity eliminates the anti-ageing effect of plasmapheresis on the liver [89]. Exosomes from young serum significantly downregulated senescence-related genes (cyclin-dependent kinase inhibitor 2A, mechanistic target of rapamycin, and insulin-like growth factor 1 receptor) and upregulated telomerase related genes (e.g., Men1, Mre11a, Tep1, Terf2, Tert, and Tnks) in lung and liver by reversing mmu-miR-126b-5p levels of aged mice [90]. Thus, circulating anti-ageing factors are not unique and may work together through different pathways to exert anti-ageing effects. Exercise stimulates the liver to produce glycosylphosphatidylinositol-specific phospholipase D (GPLD1) [21]. It cannot cross the blood–brain barrier; instead, it improves age-related cognitive decline by reducing inflammation and increasing blood supply to the brain [21]. Thus, remote mediating mechanisms between organs can be both directly and indirectly anti-ageing. Not limited to organs such as the liver, kidney and brain, the cytokines MCP-1 and IL-6 (pro-inflammatory) were found to be reduced in visceral adipose tissue (VAT) of aged (18 months) mice by exposure to young plasma (from 3-month-old mice). Ageing adipose tissue-derived stromovascular fraction cells showed a decrease in the expression of the senescence markers (p16Ink4a and p21Waf1/Cip1) [91]. Thus, amelioration of ageing-related hypofunction by providing a rejuvenating microenvironment (plasma proteome alteration) for senescent cells is widely applicable in a wide range of tissues [18].
In addition to blood-based evidence, this “youthful secretory phenotype (YSP)” (secreted by young cells) anti-ageing phenomenon is also widely observed in a variety of tissues (e.g., muscle, adipose, etc.). Cardiac stem cells are absent from the adult myocardium, but paracrine effects derived from young cardiomyocytes lengthen the telomeres not restricted to senescent cardiomyocytes. In aged rats treated with cardiac sphere-derived cells (CDCs) from young donors, circulating levels of the inflammatory cytokines interleukin-1β and interleukin-6 were reduced, along with elevated anti-inflammatory interleukin-10 levels, correlated with the observed improvements in exercise capacity, muscle reduction, hair regeneration, and renal function [92]. This research shows that young source tissue cells deliberately produce systemic benefits through systemic improvements rather than local improvements in single-organ ageing alone. Allogenic CDC intracoronary infusion in patients with heart attack increased left ventricle (LV) volume and N-terminal prob-type natriuretic peptide (NT-proBNP) compared with placebo but did not reduce scarring [93].
Further studies have shown that the ageing bone marrow can be reconstituted by tail vein transplantation of juvenile-derived antigen 1 positive (Sca-1+) bone marrow (BM) stem cells. It also promotes the rejuvenation of the heart by activating the c-x-c motif chemokine ligand 12 c-x-c chemokine receptor type 4 (Cxcl12/Cxcr4) pathway in cardiac endothelial cells [94]. CDC transplanted into the rat heart after myocardial infarction can derive exosomes into the bloodstream to promote myocardial repair via microRNAs associated with myocardial recovery [95].
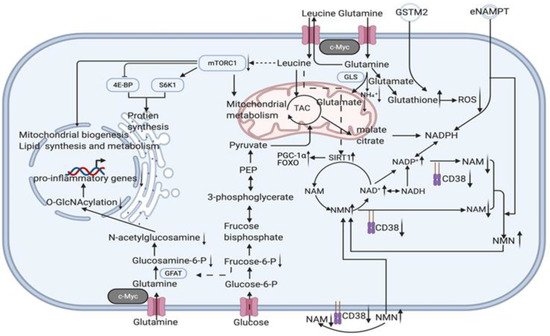
Nicotinamide adenine dinucleotide (NAD) is a metabolic cofactor that decreases with age and supplementation with its precursors, most commonly nicotinamide (NAM), is thought to be anti-ageing. Although NAM is a NAD+ precursor, NAM inhibits the NAM salvage pathway without producing a net elevation in the NAD metabolome. NAM treatment increases global protein acetylation, indicating that total sirtuins are inhibited. Combined with the reduction in nicotinamide phosphoribosyltransferase (NAMPT) levels it causes [96], the upper limit of the effect of NAD+ precursor supplementation alone may not be sufficient to meet anti-ageing requirements (enzymatic reaction balance may have an inhibitory effect on enzymes and enzyme depletion and inactivation due to ageing). Therefore, the reduction in NAD depletion caused by heavy braiding, enzyme renewal, and mitochondrial rejuvenation may be more advantageous. The NAD+-dependent deacetylase (SIRT1) in mammalian adipocytes deacetylates intracellular nicotinamide phosphoribosyltransferase (iNAMPT) to facilitate secretion to form extracellular nicotinamide phosphoribosyltransferase (eNAMPT) and enhance eNAMPT activity. eNAMPT in adipose tissue enhances hypothalamic NAD+/SIRT1 signalling and physical activity [57]. On the other hand, the hypothalamus is thought to be the superior control centre for ageing in mammals. By influencing endocrine regulatory centres, eNAMPT links adipose reprogramming anti-ageing strategies to endocrine regulation. The levels of eNAMPT in extracellular vesicles (EVs) decreased significantly with age and increased the secretion of eNAMPT-containing EVs from adipocytes; it significantly improved the behavioural phenotype of ageing and prolonged lifespan in mice [49]. In addition to the direct effects on cellular energy metabolism and redox, we should also consider the indirect effects caused by eNAMPT, such as acetylation, which is widely involved in a variety of metabolic and chromatin regulations (histone modifications) and thus extensively affects mitochondrial metabolic remodelling and epigenetic remodelling in reprogramming strategies. Some easily overlooked mechanisms of gene damage also need to be investigated in an integrated manner to complete the network of crucial factors for reversal of ageing; e.g., in addition to ROS-dependent DDR, DNA-protein cross-linking (DPC) is also one of the triggers of nuclear senescence. Gene damage induces ATM/ATR activation, which activates the deubiquitinating enzyme VCPIP1/VCIP135 by phosphorylation, and VCPIP1 deubiquitinates SPRTN(a DNA-dependent metalloproteinase) to create the conditions for its subsequent acetylation, which eventually localizes SPRTN to chromatin damage sites to cleave DPCs proteins and protect genomic stability from senescence [97]. Thus eNAMPT and the response induced by changes in NAD+/NADH ratios (and even changes in sirtuin1,3) are by no means limited to the previously described crosstalk mechanisms between ROS-induced DNA damage response (DDR) and mitochondrial dysfunction-associated senescence (MiDAS) [3,4] but are instead more broad. Reduced glutamine levels in lipid cells reduce uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) levels via glycolysis, reducing O-linked β-N-acetylglucosamine (O-GlcNAc). This ultimately leads to chronic inflammation development by promoting a pro-inflammatory transcriptional response through O-GlcNAcy of chromatin-binding proteins near inflammatory genes [48] (Figure 3)
This entry is adapted from the peer-reviewed paper 10.3390/cells11050830