The HIV-1 integrase enzyme (IN) plays a critical role in the viral life cycle by integrating the reverse-transcribed viral DNA into the host chromosome. This function of IN has been well studied, and the knowledge gained has informed the design of small molecule inhibitors that now form key components of antiretroviral therapy regimens. Recent discoveries unveiled that IN has an under-studied yet equally vital second function in HIV-1 replication. This involves IN binding to the viral RNA genome in virions, which is necessary for proper virion maturation and morphogenesis. Herein we describe these two functions of IN within the context of the HIV-1 life cycle, how IN binding to the viral genome is coordinated by the major structural protein, Gag, and discuss the value of targeting the second role of IN in virion morphogenesis.
- HIV-1
- integrase
- maturation
- integrase-RNA interactions
- protein-RNA interactions
1. Introduction
Human immunodeficiency virus type 1 (HIV-1) is the causative agent of AIDS, and since its discovery in 1983 [1][2] has become one of the leading causes of the death worldwide due to infectious disease. Intensive study of the HIV-1 life cycle has led to the identification of viral enzymes essential for virus replication, and antiretroviral compounds that specifically inhibit the functions of these enzymes have transformed HIV-1 infection from a death sentence into a manageable disease. The HIV-1 integrase enzyme (IN) plays a vital role in the viral life cycle by catalyzing the integration of viral DNA into the host chromosome. This function has been successfully targeted by a class of antiretrovirals known as integrase strand-transfer inhibitors (INSTIs) [3]. Four FDA-approved INSTIs, raltegravir [4], elvitegravir [5], dolutegravir [6], and bictegravir [7], have become key components of anti-retroviral therapy regimens and are both highly effective and well tolerated ([8][9][10], reviewed in [11]). A fifth, cabotegravir [12], is currently in late stage clinical trials. However, despite high barriers with the second-generation INSTIs, treatment does select for drug resistance [13][14][15], and mutations conferring resistance to multiple INSTIs have been reported in clinical settings [16][17], highlighting the need for continued research and development of both improved and novel antiretroviral compounds.
It was recently discovered that IN has a second essential role in the HIV-1 life cycle. IN binds the viral RNA (vRNA) genome in virions and is necessary for the proper placement of vRNA within the viral capsid lattice during virion maturation [18]. Loss of IN–RNA binding leads to mislocalization of the viral genome in virions and prevents viral replication in target cells [18]. This discovery opens up new avenues for therapeutic targeting of the second function of IN that is independent of its already targeted catalytic function.
2. Overview of the HIV-1 Life Cycle
The HIV-1 life cycle can be broadly divided into an early stage (up to integration) and a late stage (after integration). Mature HIV-1 virions consist of two copies of a single-stranded RNA genome and replicative enzymes (i.e., reverse transcriptase (RT) and IN) encased in a conical protein lattice made up by the viral capsid (CA) protein, together forming the viral “core” (Figure 1). The viral genome inside the core exists in the form of a viral ribonucleoprotein complex (vRNP) bound and condensed by the viral nucleocapsid (NC) protein, and associated with RT and IN enzymes [19]. The viral core itself is enclosed within the viral lipid envelope derived from the host cell plasma membrane. The surface of the virion is studded with the viral envelope (Env) glycoprotein trimers (reviewed in [20]). During viral entry, the Env glycoproteins engage the CD4 receptor and CXCR4/CCR5 coreceptors on the target cell, which induces a series of conformational changes in Env, resulting in membrane fusion and release of the viral core into the cytoplasm (reviewed in [21][22]).
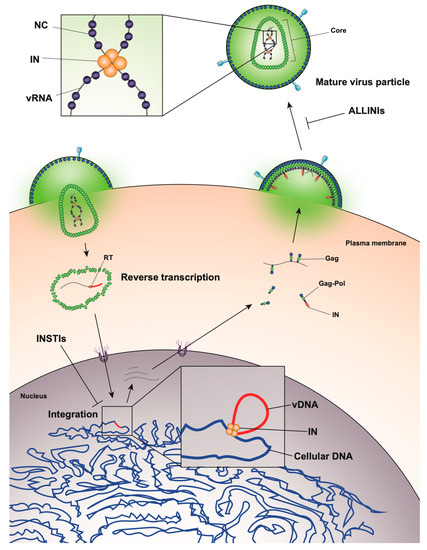
Figure 1. Overview of the human immunodeficiency virus type 1 (HIV-1) life cycle highlighting the key role of HIV-1 integrase enzyme (IN) at early and late stages of replication.
After entry, the viral core is transported towards the nucleus along microtubules [23][24] and reverse transcription ensues (Figure 1). During this stage, the core undergoes an uncoating process in which the capsid disassembles and CA monomers are shed from the lattice (reviewed in [25]). While uncoating and reverse transcription have long been thought to occur in the cytoplasm, recent studies have provided evidence that these processes are not completed until after nuclear entry [26][27]. Notwithstanding, the timing and degree of uncoating is critical for completion of reverse transcription. Several mutations in CA can destabilize the CA lattice, resulting in severe defects in reverse transcription [28][29][30][31], presumably due to premature degradation of the core components, including IN [32]. IN remains associated with the reverse transcription complex, and following completion of cDNA synthesis, a multimer of IN binds to both ends of the linear viral DNA to form the intasome, or the stable synaptic complex. The reverse transcription complex is actively transported into the nucleus through the nuclear pore complexes involving Nup 358, Nup 153, as well as CPSF6 (reviewed in [33][34]), and upon entering the nucleus IN catalyzes the integration of the viral DNA into the host cell chromosome (reviewed in [35]).
After the viral DNA is integrated into the host chromosome it serves as a template from which single full-length viral mRNA is transcribed by the host RNA polymerase II machinery (reviewed in [36], Figure 1). This viral transcript can remain unspliced or undergo a complex series of splicing events (reviewed in [36]). Fully spliced HIV-1 mRNAs code for the regulatory Tat, Rev, and Nef proteins and are exported from the nucleus via the NXF1/NXT1 pathway (reviewed in [37][38][39]). Partially spliced mRNAs code for the viral envelope Env and accessory proteins Vif, Vpr, and Vpu, while the unspliced full-length HIV-1 mRNAs can be packaged into virions as the genomic RNA or translated to generate the major structural protein, Gag, and the frameshifting variant Gag–Pol polyprotein [40][41], which additionally codes for the replicative enzymes protease (PR), RT, and IN (reviewed in [37][38][39]). Both partially spliced and unspliced HIV-1 RNAs are retained in the nucleus until they can be exported by the viral Rev protein [42][43] through a CRM1-dependent pathway [44][45][46].
Unspliced dimeric vRNA is trafficked to the plasma membrane by Gag, and this complex subsequently nucleates the assembly of nascent virions (reviewed in [47][48][49], Figure 1). During this process, the Gag and Gag–Pol polyproteins polymerize around the vRNA, acquire Env glycoproteins recruited to the budding site, and virions bud off from the infected cell in an immature state. During or shortly after budding, the virion undergoes a maturation process in which the Gag and Gag–Pol polyproteins are cleaved into separate mature proteins by the virally encoded PR enzyme. This triggers a structural rearrangement within the virion, whereby the cleaved NC proteins condense the vRNA together with RT and IN to form the vRNP, the viral CA lattice assembles around the vRNP, and the now mature virion is ready to infect a new target cell and reinitiate the viral life cycle (reviewed in [47][48]).
3. Role of Integrase in Particle Morphogenesis
While integration is the canonical function of IN, early mutagenesis studies indicated that IN may also play a role in other aspects of virus replication. In particular, a group of IN substitutions referred to as class II IN mutations lead to pleiotropic effects in HIV-1 replication, including defects in particle assembly [50][51][52][53][54][55][56][57][58][59][60][61][62][63], morphogenesis [18][50][53][59][60][61][64][65], and reverse transcription in target cells [18][66][50][55][57][58][59][61][63][64][65][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82], in some cases without impacting IN catalytic function in vitro [83][53][54][57][58][68][69][72][74][84][85]. When visualized using electron microscopy, class II IN mutant viruses generate particles with vRNP complexes mislocalized outside the capsid lattice [18][50][53][59][60][61][64][65]. A similar phenotype was noted in IN-deleted viruses [60], again suggesting that IN is necessary for proper virion morphogenesis. Such aberrant viral particles are generally referred to as “eccentric particles,” due to the mislocalization of the vRNPs outside the capsid lattice, and are morphologically distinct from immature virions.
Surprisingly, it was recently discovered that treatment of virus-producing cells with a class of 2-(quinolin-3-yl) acetic acid derivatives known as allosteric IN inhibitors (ALLINIs) (also called noncatalytic IN inhibitors (NCINIs), lens epithelium-derived growth factor (LEDGF)/p75-IN inhibitors (LEDGINs), IN-LEDGF/p75 allosteric inhibitors (INLAIs), or multimeric IN inhibitors (MINIs)) results in generation of particles with eccentric morphologies [64][65][86][87]. ALLINIs were originally designed to prevent integration by interfering with IN binding to the cellular cofactor, lens epithelium-derived growth factor (LEDGF/p75), important for targeting the viral pre-integration complex to the host chromosome [88]. The compounds compete with LEDGF binding to IN by engaging the V-shaped binding pocket created by the catalytic core domain of two IN dimers in the intasome complex [65][88][89][90][91][92][93]. In addition to preventing IN–LEDGF interactions, ALLINIs also prevent integration in a LEDGF-independent manner by inducing aberrant IN multimerization, locking IN in catalytically inactive multimers that are unable to assemble on viral DNA and carry out the integration reaction (reviewed in [89][93]). However, subsequent studies found that many ALLINIs are more potent when added to producer cells, and that they inhibit viral replication at the later stages of the viral life cycle [65][86][87][91][92]. Specifically, treatment with ALLINIs interferes with virion morphogenesis and leads to the generation of eccentric viral particles with vRNPs mislocalized outside the capsid lattice, strikingly similar to those generated by class II IN mutations [65][86][87][91][94]. Similar to the mechanism by which they can prevent integration, ALLINIs are proposed to interfere with virion morphogenesis by inducing aberrant IN multimerization, and mutations that confer resistance to ALLINIs also prevent ALLINI-induced IN multimerization [86][95]. Many class II IN mutations also alter IN multimerization [96][84][97][98], suggesting that proper multimerization is important for IN’s function during virion morphogenesis. However, a defined mechanism by which IN ensures viral RNA is correctly packaged inside the capsid lattice remained elusive for many years.
A seminal study in 2016 revealed that IN binds viral genomic RNA in mature virions, and that IN–RNA binding is necessary for viral replication [18]. Crosslinking immunoprecipitation sequencing (CLIP-seq), an approach that captures protein–RNA interactions in relevant physiological settings [99], was instrumental in this discovery and demonstrated that IN binds the HIV-1 genome at discrete sites with a distinct binding pattern from that of NC. IN not only binds RNA, but also modulates RNA structure in vitro by bridging multiple RNA molecules together [18]. Several basic residues in the IN CTD, K264, K266, and K273, directly interact with RNA, and substitutions at these positions abolish IN–RNA binding in virions. Importantly, virus production in the presence of ALLINIs, BI-D and BI-B2, also prevented IN–RNA binding, likely through aberrant IN multimerization as detailed below [18]. Finally, inhibiting IN interactions with RNA, either by introducing mutations at the CTD binding site or by ALLINI treatment, leads to the generation of eccentric, non-infectious viral particles (Figure 2) with vRNPs mislocalized outside of the core [18].
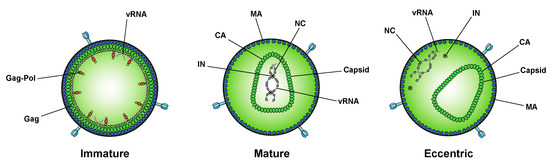
Figure 2. Inhibition of IN–RNA interactions result in the formation of eccentric virions. Immature viral particles consist of many molecules of Gag and Gag–Pol concentrically arranged along the inner leaflet of the viral membrane and bound to viral RNA (vRNA) at the NC domain. In mature viral particles, the vRNA is bound by NC and condensed with RT and IN to form the vRNP, which is enclosed in the conical capsid lattice made up of CA monomers. In eccentric viral particles, as observed upon inhibition of IN binding to the viral genome, the vRNP is mislocalized outside of the capsid lattice.
This discovery uncovered an important clue as to how IN contributes to virion morphogenesis by highlighting its interaction with RNA, but also raised questions regarding the relationship between IN–RNA binding, IN multimerization, and proper virion morphogenesis. Specifically, the findings described above strongly indicated that IN–RNA binding was a driving force behind proper virion maturation, but the observation that numerous class II IN mutations lead to eccentric virion morphology, yet are distally located from the RNA-binding site in the IN CTD, argued that the loss of IN–RNA may be correlative and another characteristic of IN, such as its proper multimerization, was the true determining factor.
Recent characterization of the effects of class II IN mutations on IN–RNA binding, IN multimerization, and virion morphology has revealed that all of the class II IN substitutions examined compromise IN–RNA binding and lead to the generation of eccentric viral particles [94]. IN–RNA binding was prevented by one of three distinct mechanisms: reducing IN levels in virions and precluding formation of IN–RNA complexes, directly preventing IN–RNA binding without substantially affecting IN levels or IN multimerization, but most commonly through adversely affecting functional IN multimerization and indirectly impairing IN–RNA binding. In vitro, IN binds RNA as tetramers, and class II IN mutant proteins that form predominantly dimers have a reduced affinity for RNA. The mutations cause an even greater defect in the ability of IN to bridge multiple RNA molecules, suggesting that although IN dimers may be able to weakly bind RNA, IN must form tetramers to bind RNA with high affinity and recruit additional viral RNA molecules, as would possibly occur when viral RNA is condensed and placed within the core during maturation. Taken together, these recent findings argue that proper IN multimerization is likely a prerequisite for IN–RNA binding in virions and is important to IN’s function in virion morphogenesis. The identification of multiple mechanisms responsible for the loss of IN–RNA binding helps answer how multiple IN substitutions can cause the same phenotype and strengthens the conclusion that IN–RNA interactions account for the key role of IN in virion morphogenesis [94].
Whether IN plays a similar role during virion maturation in other retroviruses has yet to be studied. Although the process of maturation is relatively conserved across different retroviruses, there is considerable diversity in particle and core morphologies [100][101][102]. Interestingly, mutations in the C-terminus of the murine leukemia virus (MLV) IN cause defects in reverse transcription reminiscent of those caused by class II IN mutations in HIV-1 [103][104]. More studies are needed to determine if this defect is the result of aberrant virion morphology, or if IN–RNA interaction is required for proper virion morphology.
4. Therapeutic Outlook and Conclusions
Currently, all clinically approved IN inhibitors share a common mode of action and target the strand-transfer reaction during integration, making viral cross-resistance a problem. Therefore, there has long been interest in targeting alternative functions of IN, and its vital role during virion morphogenesis has become an attractive drug target. While not initially designed to interfere with viral particle maturation, many ALLINIs lead to the generation of eccentric viral particles in addition to inhibiting integration [18]. This dual-mode of action allows these compounds to retain antiviral activity even when viral resistance develops to one mode of action [105], and gives ALLINIs a distinct and non-overlapping resistance profile with INSTIs [106][107]. While resistance mutations do arise to ALLINIs, they are accompanied by a viral fitness cost. Importantly, mutations that confer resistance to ALLINIs can disrupt virion morphogenesis themselves, and compensatory mutations are required to overcome this defect [108], indicating a high barrier to resistance. Several compounds are currently in clinical trials after demonstrating favorable bioavailability, tolerability, and pharmacokinetics, and with more in development, ALLINIs are a promising new class of antiretrovirals.
CA has also emerged as a potential target for compounds designed to interfere with virion morphogenesis. Compounds that disrupt the stability of the capsid lattice and cause morphological defects in virions also block viral replication at or prior to reverse transcription [109][110], and they likely lead to the premature loss of vRNA and IN similar to how ALLINIs work. Two recent studies reported the effectiveness of a CA-targeting small molecule compounds in vivo, both in a humanized mouse model [111] and in humans [112]. Both compounds demonstrated potent and sustained antiviral activity in vitro, with no measurable cross-resistance with other antiretroviral drugs [112]. While it is important to note that these compounds led to morphological aberrations in virions by accelerating CA assembly rather than destabilizing it and appear to exert their antiviral effects primarily by preventing the nuclear import of viral DNA, they still highlight the importance of proper capsid assembly to viral replication and the value of interfering with virion morphogenesis as a therapeutic strategy.
In conclusion, virion morphogenesis is a critical step in the HIV-1 life cycle, and the discovery that IN plays a key role in this process opens up new doors for therapeutic interventions. IN is crucial for ensuring that the viral RNA genome is packaged inside of the capsid lattice in virions, and interfering with this function of IN leads to morphological defects that prevent further viral replication. Small molecule compounds that exert this effect can complement existing antiretroviral compounds already in the clinic, and when used in combination with INSTIs could further raise the barrier to drug resistance. Destabilizing the viral capsid by targeting CA has similar effects on viral replication, and is also a viable therapeutic strategy. A better understanding of the events surrounding virion morphogenesis will be important to help guide the design of future therapeutics.
This entry is adapted from the peer-reviewed paper 10.3390/v12091005
References
- Barré-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vézinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871.
- Gallo, R.C.; Salahuddin, S.Z.; Popovic, M.; Shearer, G.M.; Kaplan, M.; Haynes, B.F.; Palker, T.J.; Redfield, R.; Oleske, J.; Safai, B.; et al. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 1984, 224, 500–503.
- Hazuda, D.J.; Felock, P.; Witmer, M.; Wolfe, A.; Stillmock, K.; Grobler, J.A.; Espeseth, A.; Gabryelski, L.; Schleif, W.; Blau, C.; et al. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000, 287, 646–650.
- Summa, V.; Petrocchi, A.; Bonelli, F.; Crescenzi, B.; Donghi, M.; Ferrara, M.; Fiore, F.; Gardelli, C.; Gonzalez Paz, O.; Hazuda, D.J.; et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 2008, 51, 5843–5855.
- Ramanathan, S.; Mathias, A.A.; German, P.; Kearney, B.P. Clinical pharmacokinetic and pharmacodynamic profile of the HIV integrase inhibitor elvitegravir. Clin. Pharm. 2011, 50, 229–244.
- Min, S.; Song, I.; Borland, J.; Chen, S.; Lou, Y.; Fujiwara, T.; Piscitelli, S.C. Pharmacokinetics and safety of S/GSK1349572, a next-generation HIV integrase inhibitor, in healthy volunteers. Antimicrob. Agents Chemother. 2010, 54, 254–258.
- Tsiang, M.; Jones, G.S.; Goldsmith, J.; Mulato, A.; Hansen, D.; Kan, E.; Tsai, L.; Bam, R.A.; Stepan, G.; Stray, K.M.; et al. Antiviral Activity of Bictegravir (GS-9883), a Novel Potent HIV-1 Integrase Strand Transfer Inhibitor with an Improved Resistance Profile. Antimicrob. Agents Chemother. 2016, 60, 7086–7097.
- Rockstroh, J.K.; DeJesus, E.; Lennox, J.L.; Yazdanpanah, Y.; Saag, M.S.; Wan, H.; Rodgers, A.J.; Walker, M.L.; Miller, M.; DiNubile, M.J.; et al. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatment-naive HIV-1-infected patients: Final 5-year results from STARTMRK. J. Acquir. Immune Defic. Syndr. 2013, 63, 77–85.
- Taha, H.; Das, A.; Das, S. Clinical effectiveness of dolutegravir in the treatment of HIV/AIDS. Infect. Drug Resist. 2015, 8, 339–352.
- Smith, S.J.; Zhao, X.Z.; Passos, D.O.; Lyumkis, D.; Burke, T.R.; Hughes, S.H. HIV-1 Integrase Inhibitors that are active against Drug-Resistant Integrase Mutants. Antimicrob. Agents Chemother. 2020, 62, e01035-18.
- Brooks, K.M.; Sherman, E.M.; Egelund, E.F.; Brotherton, A.; Durham, S.; Badowski, M.E.; Cluck, D.B. Integrase Inhibitors: After 10 Years of Experience, Is the Best Yet to Come? Pharmacotherapy 2019, 39, 576–598.
- Smith, S.J.; Zhao, X.Z.; Burke, T.R., Jr.; Hughes, S.H. Efficacies of Cabotegravir and Bictegravir against drug-resistant HIV-1 integrase mutants. Retrovirology 2018, 15, 37.
- Wijting, I.E.A.; Lungu, C.; Rijnders, B.J.A.; van der Ende, M.E.; Pham, H.T.; Mesplede, T.; Pas, S.D.; Voermans, J.J.C.; Schuurman, R.; van de Vijver, D.A.M.C.; et al. HIV-1 Resistance Dynamics in Patients with Virologic Failure to Dolutegravir Maintenance Monotherapy. J. Infect. Dis. 2018, 218, 688–697.
- Radzio-Basu, J.; Council, O.; Cong, M.E.; Ruone, S.; Newton, A.; Wei, X.; Mitchell, J.; Ellis, S.; Petropoulos, C.J.; Huang, W.; et al. Drug resistance emergence in macaques administered cabotegravir long-acting for pre-exposure prophylaxis during acute SHIV infection. Nat. Commun. 2019, 10, 2005.
- Anstett, K.; Brenner, B.; Mesplede, T.; Wainberg, M.A. HIV drug resistance against strand transfer integrase inhibitors. Retrovirology 2017, 14, 36.
- Rossouw, T.M.; Hitchcock, S.; Botes, M. The end of the line? A case of drug resistance to third-line antiretroviral therapy. S. Afr. J. HIV Med. 2016, 17, 454.
- Zhang, W.W.; Cheung, P.K.; Oliveira, N.; Robbins, M.A.; Harrigan, P.R.; Shahid, A. Accumulation of Multiple Mutations In Vivo Confers Cross-Resistance to New and Existing Integrase Inhibitors. J. Infect. Dis. 2018, 218, 1773–1776.
- Kessl, J.J.; Kutluay, S.B.; Townsend, D.; Rebensburg, S.; Slaughter, A.; Larue, R.C.; Shkriabai, N.; Bakouche, N.; Fuchs, J.R.; Bieniasz, P.D.; et al. HIV-1 Integrase Binds the Viral RNA Genome and Is Essential during Virion Morphogenesis. Cell 2016, 166, 1257–1268.
- Mattei, S.; Schur, F.K.; Briggs, J.A. Retrovirus maturation-an extraordinary structural transformation. Curr. Opin. Virol. 2016, 18, 27–35.
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 2011, 410, 582–608.
- Wang, Q.; Finzi, A.; Sodroski, J. The Conformational States of the HIV-1 Envelope Glycoproteins. Trends Microbiol. 2020, 28, 655–667.
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891.
- Dharan, A.; Opp, S.; Abdel-Rahim, O.; Keceli, S.K.; Imam, S.; Diaz-Griffero, F.; Campbell, E.M. Bicaudal D2 facilitates the cytoplasmic trafficking and nuclear import of HIV-1 genomes during infection. Proc. Natl. Acad. Sci. USA 2017, 114, E10707–E10716.
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 2002, 159, 441–452.
- Campbell, E.M.; Hope, T.J. HIV-1 capsid: The multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 2015, 13, 471–483.
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E.M. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 2020, 5, 1088–1095.
- Burdick, R.C.; Li, C.; Munshi, M.; Rawson, J.M.O.; Nagashima, K.; Hu, W.S.; Pathak, V.K. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493.
- Fitzon, T.; Leschonsky, B.; Bieler, K.; Paulus, C.; Schroder, J.; Wolf, H.; Wagner, R. Proline residues in the HIV-1 NH2-terminal capsid domain: Structure determinants for proper core assembly and subsequent steps of early replication. Virology 2000, 268, 294–307.
- Forshey, B.M.; von Schwedler, U.; Sundquist, W.I.; Aiken, C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 2002, 76, 5667–5677.
- Reicin, A.S.; Ohagen, A.; Yin, L.; Hoglund, S.; Goff, S.P. The role of Gag in human immunodeficiency virus type 1 virion morphogenesis and early steps of the viral life cycle. J. Virol. 1996, 70, 8645–8652.
- Tang, S.; Murakami, T.; Agresta, B.E.; Campbell, S.; Freed, E.O.; Levin, J.G. Human immunodeficiency virus type 1 N-terminal capsid mutants that exhibit aberrant core morphology and are blocked in initiation of reverse transcription in infected cells. J. Virol. 2001, 75, 9357–9366.
- Eschbach, J.E.; Elliott, J.L.; Li, W.; Zadrozny, K.K.; Davis, K.; Mohammed, S.; Lawson, D.Q.; Pornillos, O.; Engelman, A.N.; Kutluay, S.B. Capsid lattice destabilization leads to premature loss of the viral genome and integrase enzyme during HIV-1 infection. Biorxiv 2020.
- Bhargava, A.; Lahaye, X.; Manel, N. Let me in: Control of HIV nuclear entry at the nuclear envelope. Cytokine Growth Factor Rev. 2018, 40, 59–67.
- Novikova, M.; Zhang, Y.; Freed, E.O.; Peng, K. Multiple Roles of HIV-1 Capsid during the Virus Replication Cycle. Virol. Sin. 2019, 34, 119–134.
- Lesbats, P.; Engelman, A.N.; Cherepanov, P. Retroviral DNA Integration. Chem. Rev. 2016, 116, 12730–12757.
- Karn, J.; Stoltzfus, C.M. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb. Perspect. Med. 2012, 2, a006916.
- Harris, M.E.; Hope, T.J. RNA export: Insights from viral models. Essays Biochem. 2000, 36, 115–127.
- Cullen, B.R. Nuclear mRNA export: Insights from virology. Trends Biochem. Sci. 2003, 28, 419–424.
- Wodrich, H.; Krausslich, H.G. Nucleocytoplasmic RNA transport in retroviral replication. Results Probl. Cell Differ. 2001, 34, 197–217.
- Jacks, T.; Power, M.D.; Masiarz, F.R.; Luciw, P.A.; Barr, P.J.; Varmus, H.E. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 1988, 331, 280–283.
- Wilson, W.; Braddock, M.; Adams, S.E.; Rathjen, P.D.; Kingsman, S.M.; Kingsman, A.J. HIV expression strategies: Ribosomal frameshifting is directed by a short sequence in both mammalian and yeast systems. Cell 1988, 55, 1159–1169.
- Malim, M.H.; Cullen, B.R. HIV-1 structural gene expression requires the binding of multiple Rev monomers to the viral RRE: Implications for HIV-1 latency. Cell 1991, 65, 241–248.
- Malim, M.H.; Tiley, L.S.; McCarn, D.F.; Rusche, J.R.; Hauber, J.; Cullen, B.R. HIV-1 structural gene expression requires binding of the Rev trans-activator to its RNA target sequence. Cell 1990, 60, 675–683.
- Pollard, V.W.; Malim, M.H. The HIV-1 Rev protein. Annu. Rev. Microbiol. 1998, 52, 491–532.
- Fornerod, M.; Ohno, M.; Yoshida, M.; Mattaj, I.W. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 1997, 90, 1051–1060.
- Fukuda, M.; Asano, S.; Nakamura, T.; Adachi, M.; Yoshida, M.; Yanagida, M.; Nishida, E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 1997, 390, 308–311.
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924.
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484–496.
- Bell, N.M.; Lever, A.M. HIV Gag polyprotein: Processing and early viral particle assembly. Trends Microbiol. 2013, 21, 136–144.
- Engelman, A.; Englund, G.; Orenstein, J.M.; Martin, M.A.; Craigie, R. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 1995, 69, 2729–2736.
- Ansari-Lari, M.A.; Donehower, L.A.; Gibbs, R.A. Analysis of human immunodeficiency virus type 1 integrase mutants. Virology 1995, 213, 680.
- Bukovsky, A.; Gottlinger, H. Lack of integrase can markedly affect human immunodeficiency virus type 1 particle production in the presence of an active viral protease. J. Virol. 1996, 70, 6820–6825.
- Jenkins, T.M.; Engelman, A.; Ghirlando, R.; Craigie, R. A soluble active mutant of HIV-1 integrase: Involvement of both the core and carboxyl-terminal domains in multimerization. J. Biol. Chem. 1996, 271, 7712–7718.
- Kalpana, G.V.; Reicin, A.; Cheng, G.S.; Sorin, M.; Paik, S.; Goff, S.P. Isolation and characterization of an oligomerization-negative mutant of HIV-1 integrase. Virology 1999, 259, 274–285.
- Leavitt, A.D.; Robles, G.; Alesandro, N.; Varmus, H.E. Human immunodeficiency virus type 1 integrase mutants retain in vitro integrase activity yet fail to integrate viral DNA efficiently during infection. J. Virol. 1996, 70, 721–728.
- Liao, W.H.; Wang, C.T. Characterization of human immunodeficiency virus type 1 Pr160 gag-pol mutants with truncations downstream of the protease domain. Virology 2004, 329, 180–188.
- Lu, R.; Ghory, H.Z.; Engelman, A. Genetic analyses of conserved residues in the carboxyl-terminal domain of human immunodeficiency virus type 1 integrase. J. Virol. 2005, 79, 10356–10368.
- Lu, R.; Limon, A.; Devroe, E.; Silver, P.A.; Cherepanov, P.; Engelman, A. Class II integrase mutants with changes in putative nuclear localization signals are primarily blocked at a postnuclear entry step of human immunodeficiency virus type 1 replication. J. Virol. 2004, 78, 12735–12746.
- Nakamura, T.; Masuda, T.; Goto, T.; Sano, K.; Nakai, M.; Harada, S. Lack of infectivity of HIV-1 integrase zinc finger-like domain mutant with morphologically normal maturation. Biochem. Biophys. Res. Commun. 1997, 239, 715–722.
- Quillent, C.; Borman, A.M.; Paulous, S.; Dauguet, C.; Clavel, F. Extensive regions of pol are required for efficient human immunodeficiency virus polyprotein processing and particle maturation. Virology 1996, 219, 29–36.
- Shin, C.G.; Taddeo, B.; Haseltine, W.A.; Farnet, C.M. Genetic analysis of the human immunodeficiency virus type 1 integrase protein. J. Virol. 1994, 68, 1633–1642.
- Taddeo, B.; Haseltine, W.A.; Farnet, C.M. Integrase mutants of human immunodeficiency virus type 1 with a specific defect in integration. J. Virol. 1994, 68, 8401–8405.
- Wu, X.; Liu, H.; Xiao, H.; Conway, J.A.; Hehl, E.; Kalpana, G.V.; Prasad, V.; Kappes, J.C. Human immunodeficiency virus type 1 integrase protein promotes reverse transcription through specific interactions with the nucleoprotein reverse transcription complex. J. Virol. 1999, 73, 2126–2135.
- Fontana, J.; Jurado, K.A.; Cheng, N.; Ly, N.L.; Fuchs, J.R.; Gorelick, R.J.; Engelman, A.N.; Steven, A.C. Distribution and Redistribution of HIV-1 Nucleocapsid Protein in Immature, Mature, and Integrase-Inhibited Virions: A Role for Integrase in Maturation. J. Virol. 2015, 89, 9765–9780.
- Jurado, K.A.; Wang, H.; Slaughter, A.; Feng, L.; Kessl, J.J.; Koh, Y.; Wang, W.; Ballandras-Colas, A.; Patel, P.A.; Fuchs, J.R.; et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. USA 2013, 110, 8690–8695.
- Engelman, A. In vivo analysis of retroviral integrase structure and function. Adv. Virus Res. 1999, 52, 411–426.
- Ao, Z.; Fowke, K.R.; Cohen, E.A.; Yao, X. Contribution of the C-terminal tri-lysine regions of human immunodeficiency virus type 1 integrase for efficient reverse transcription and viral DNA nuclear import. Retrovirology 2005, 2, 62.
- Busschots, K.; Voet, A.; De Maeyer, M.; Rain, J.C.; Emiliani, S.; Benarous, R.; Desender, L.; Debyser, Z.; Christ, F. Identification of the LEDGF/p75 binding site in HIV-1 integrase. J. Mol. Biol. 2007, 365, 1480–1492.
- Engelman, A.; Liu, Y.; Chen, H.; Farzan, M.; Dyda, F. Structure-based mutagenesis of the catalytic domain of human immunodeficiency virus type 1 integrase. J. Virol. 1997, 71, 3507–3514.
- Limon, A.; Devroe, E.; Lu, R.; Ghory, H.Z.; Silver, P.A.; Engelman, A. Nuclear localization of human immunodeficiency virus type 1 preintegration complexes (PICs): V165A and R166A are pleiotropic integrase mutants primarily defective for integration, not PIC nuclear import. J. Virol. 2002, 76, 10598–10607.
- Lloyd, A.G.; Ng, Y.S.; Muesing, M.A.; Simon, V.; Mulder, L.C. Characterization of HIV-1 integrase N-terminal mutant viruses. Virology 2007, 360, 129–135.
- Lu, R.; Vandegraaff, N.; Cherepanov, P.; Engelman, A. Lys-34, dispensable for integrase catalysis, is required for preintegration complex function and human immunodeficiency virus type 1 replication. J. Virol. 2005, 79, 12584–12591.
- Masuda, T.; Planelles, V.; Krogstad, P.; Chen, I.S. Genetic analysis of human immunodeficiency virus type 1 integrase and the U3 att site: Unusual phenotype of mutants in the zinc finger-like domain. J. Virol. 1995, 69, 6687–6696.
- Rahman, S.; Lu, R.; Vandegraaff, N.; Cherepanov, P.; Engelman, A. Structure-based mutagenesis of the integrase-LEDGF/p75 interface uncouples a strict correlation between in vitro protein binding and HIV-1 fitness. Virology 2007, 357, 79–90.
- Riviere, L.; Darlix, J.L.; Cimarelli, A. Analysis of the viral elements required in the nuclear import of HIV-1 DNA. J. Virol. 2010, 84, 729–739.
- Tsurutani, N.; Kubo, M.; Maeda, Y.; Ohashi, T.; Yamamoto, N.; Kannagi, M.; Masuda, T. Identification of critical amino acid residues in human immunodeficiency virus type 1 IN required for efficient proviral DNA formation at steps prior to integration in dividing and nondividing cells. J. Virol. 2000, 74, 4795–4806.
- Wiskerchen, M.; Muesing, M.A. Human immunodeficiency virus type 1 integrase: Effects of mutations on viral ability to integrate, direct viral gene expression from unintegrated viral DNA templates, and sustain viral propagation in primary cells. J. Virol. 1995, 69, 376–386.
- Zhu, K.; Dobard, C.; Chow, S.A. Requirement for integrase during reverse transcription of human immunodeficiency virus type 1 and the effect of cysteine mutations of integrase on its interactions with reverse transcriptase. J. Virol. 2004, 78, 5045–5055.
- De Houwer, S.; Demeulemeester, J.; Thys, W.; Rocha, S.; Dirix, L.; Gijsbers, R.; Christ, F.; Debyser, Z. The HIV-1 integrase mutant R263A/K264A is 2-fold defective for TRN-SR2 binding and viral nuclear import. J. Biol. Chem. 2014, 289, 25351–25361.
- Johnson, B.C.; Metifiot, M.; Ferris, A.; Pommier, Y.; Hughes, S.H. A homology model of HIV-1 integrase and analysis of mutations designed to test the model. J. Mol. Biol. 2013, 425, 2133–2146.
- Mohammed, K.D.; Topper, M.B.; Muesing, M.A. Sequential deletion of the integrase (Gag-Pol) carboxyl terminus reveals distinct phenotypic classes of defective HIV-1. J. Virol. 2011, 85, 4654–4666.
- Shehu-Xhilaga, M.; Hill, M.; Marshall, J.A.; Kappes, J.; Crowe, S.M.; Mak, J. The conformation of the mature dimeric human immunodeficiency virus type 1 RNA genome requires packaging of pol protein. J. Virol. 2002, 76, 4331–4340.
- Engelman, A.; Craigie, R. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J. Virol. 1992, 66, 6361–6369.
- Lutzke, R.A.; Plasterk, R.H. Structure-based mutational analysis of the C-terminal DNA-binding domain of human immunodeficiency virus type 1 integrase: Critical residues for protein oligomerization and DNA binding. J. Virol. 1998, 72, 4841–4848.
- Lutzke, R.A.; Vink, C.; Plasterk, R.H. Characterization of the minimal DNA-binding domain of the HIV integrase protein. Nucleic Acids Res. 1994, 22, 4125–4131.
- Balakrishnan, M.; Yant, S.R.; Tsai, L.; O’Sullivan, C.; Bam, R.A.; Tsai, A.; Niedziela-Majka, A.; Stray, K.M.; Sakowicz, R.; Cihlar, T. Non-catalytic site HIV-1 integrase inhibitors disrupt core maturation and induce a reverse transcription block in target cells. PLoS ONE 2013, 8, e74163.
- Desimmie, B.A.; Schrijvers, R.; Demeulemeester, J.; Borrenberghs, D.; Weydert, C.; Thys, W.; Vets, S.; Van Remoortel, B.; Hofkens, J.; De Rijck, J.; et al. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 2013, 10, 57.
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448.
- Kessl, J.J.; Jena, N.; Koh, Y.; Taskent-Sezgin, H.; Slaughter, A.; Feng, L.; de Silva, S.; Wu, L.; Le Grice, S.F.; Engelman, A.; et al. Multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 2012, 287, 16801–16811.
- Le Rouzic, E.; Bonnard, D.; Chasset, S.; Bruneau, J.M.; Chevreuil, F.; Le Strat, F.; Nguyen, J.; Beauvoir, R.; Amadori, C.; Brias, J.; et al. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology 2013, 10, 144.
- Sharma, A.; Slaughter, A.; Jena, N.; Feng, L.; Kessl, J.J.; Fadel, H.J.; Malani, N.; Male, F.; Wu, L.; Poeschla, E.; et al. A new class of multimerization selective inhibitors of HIV-1 integrase. PLoS Pathog. 2014, 10, e1004171.
- Gupta, K.; Brady, T.; Dyer, B.M.; Malani, N.; Hwang, Y.; Male, F.; Nolte, R.T.; Wang, L.; Velthuisen, E.; Jeffrey, J.; et al. Allosteric inhibition of human immunodeficiency virus integrase: Late block during viral replication and abnormal multimerization involving specific protein domains. J. Biol. Chem. 2014, 289, 20477–20488.
- Tsiang, M.; Jones, G.S.; Niedziela-Majka, A.; Kan, E.; Lansdon, E.B.; Huang, W.; Hung, M.; Samuel, D.; Novikov, N.; Xu, Y.; et al. New class of HIV-1 integrase (IN) inhibitors with a dual mode of action. J. Biol. Chem. 2012, 287, 21189–21203.
- Elliott, J.; Eschbach, J.E.; Koneru, P.C.; Li, W.; Chavez, M.P.; Townsend, D.; Lawson, D.; Engelman, A.N.; Kvaratskhelia, M.; Kutluay, S.B. Integrase-RNA interactions underscore the critical role of integrase in HIV-1 virion morphogenesis. bioRxiv 2019.
- Feng, L.; Sharma, A.; Slaughter, A.; Jena, N.; Koh, Y.; Shkriabai, N.; Larue, R.C.; Patel, P.A.; Mitsuya, H.; Kessl, J.J.; et al. The A128T resistance mutation reveals aberrant protein multimerization as the primary mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 2013, 288, 15813–15820.
- McKee, C.J.; Kessl, J.J.; Shkriabai, N.; Dar, M.J.; Engelman, A.; Kvaratskhelia, M. Dynamic modulation of HIV-1 integrase structure and function by cellular lens epithelium-derived growth factor (LEDGF) protein. J. Biol. Chem. 2008, 283, 31802–31812.
- Hare, S.; Di Nunzio, F.; Labeja, A.; Wang, J.; Engelman, A.; Cherepanov, P. Structural basis for functional tetramerization of lentiviral integrase. PLoS Pathog. 2009, 5, e1000515.
- Pandey, K.K.; Bera, S.; Grandgenett, D.P. The HIV-1 integrase monomer induces a specific interaction with LTR DNA for concerted integration. Biochemistry 2011, 50, 9788–9796.
- Kutluay, S.B.; Zang, T.; Blanco-Melo, D.; Powell, C.; Jannain, D.; Errando, M.; Bieniasz, P.D. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell 2014, 159, 1096–1109.
- Perilla, J.R.; Gronenborn, A.M. Molecular Architecture of the Retroviral Capsid. Trends Biochem. Sci. 2016, 41, 410–420.
- Martin, J.L.; Cao, S.; Maldonado, J.O.; Zhang, W.; Mansky, L.M. Distinct Particle Morphologies Revealed through Comparative Parallel Analyses of Retrovirus-Like Particles. J. Virol. 2016, 90, 8074–8084.
- Zhang, W.; Cao, S.; Martin, J.L.; Mueller, J.D.; Mansky, L.M. Morphology and ultrastructure of retrovirus particles. Aims Biophys. 2015, 2, 343–369.
- Steinrigl, A.; Nosek, D.; Ertl, R.; Gunzburg, W.H.; Salmons, B.; Klein, D. Mutations in the catalytic core or the C-terminus of murine leukemia virus (MLV) integrase disrupt virion infectivity and exert diverse effects on reverse transcription. Virology 2007, 362, 50–59.
- Lai, L.; Liu, H.; Wu, X.; Kappes, J.C. Moloney murine leukemia virus integrase protein augments viral DNA synthesis in infected cells. J. Virol. 2001, 75, 11365–11372.
- Bonnard, D.; Le Rouzic, E.; Eiler, S.; Amadori, C.; Orlov, I.; Bruneau, J.M.; Brias, J.; Barbion, J.; Chevreuil, F.; Spehner, D.; et al. Structure-function analyses unravel distinct effects of allosteric inhibitors of HIV-1 integrase on viral maturation and integration. J. Biol. Chem. 2018, 293, 6172–6186.
- Fenwick, C.; Amad, M.; Bailey, M.D.; Bethell, R.; Bös, M.; Bonneau, P.; Cordingley, M.; Coulombe, R.; Duan, J.; Edwards, P.; et al. Preclinical profile of BI 224436, a novel HIV-1 non-catalytic-site integrase inhibitor. Antimicrob. Agents Chemother. 2014, 58, 3233–3244.
- Choi, E.; Mallareddy, J.R.; Lu, D.; Kolluru, S. Recent advances in the discovery of small-molecule inhibitors of HIV-1 integrase. Future Sci. OA 2018, 4, FSO338.
- Hoyte, A.C.; Jamin, A.V.; Koneru, P.C.; Kobe, M.J.; Larue, R.C.; Fuchs, J.R.; Engelman, A.N.; Kvaratskhelia, M. Resistance to pyridine-based inhibitor KF116 reveals an unexpected role of integrase in HIV-1 Gag-Pol polyprotein proteolytic processing. J. Biol. Chem. 2017, 292, 19814–19825.
- Wang, W.; Zhou, J.; Halambage, U.D.; Jurado, K.A.; Jamin, A.V.; Wang, Y.; Engelman, A.N.; Aiken, C. Inhibition of HIV-1 Maturation via Small-Molecule Targeting of the Amino-Terminal Domain in the Viral Capsid Protein. J. Virol. 2017, 91.
- Blair, W.S.; Pickford, C.; Irving, S.L.; Brown, D.G.; Anderson, M.; Bazin, R.; Cao, J.; Ciaramella, G.; Isaacson, J.; Jackson, L.; et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010, 6, e1001220.
- Yant, S.R.; Mulato, A.; Hansen, D.; Tse, W.C.; Niedziela-Majka, A.; Zhang, J.R.; Stepan, G.J.; Jin, D.; Wong, M.H.; Perreira, J.M.; et al. A highly potent long-acting small-molecule HIV-1 capsid inhibitor with efficacy in a humanized mouse model. Nat. Med. 2019, 25, 1377–1384.
- Link, J.O.; Rhee, M.S.; Tse, W.C.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D.; et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020.