It has been proposed that t pharmacological chaperones (PCs), can restore the folding, trafficking, and biological activity of mutated enzymes. PCs have the advantages of wide tissue distribution, potential oral administration, lower production cost, and fewer issues of immunogenicity than enzyme replacement therapy. In this paper, we will review the advances in the identification and characterization of PCs for the MPS.
- Mucopolysaccharidoses
- Morquio A
- Hunter
- Morquio B
- Protein Folding
MPS are genetic disorders caused by the deficiency of 1 of 11 lysosomal enzymes involved in the metabolism of glycosaminoglycans (GAGs). Enzyme deficiencies may lead to the accumulation of the GAGs heparan sulfate (HS), dermatan sulfate (DS), keratan sulfate (KS), chondroitin sulfate (CS), or hyaluronan within the lysosomes [[1]]. As a result, partially catabolized GAGs accumulate in the lysosomes of several tissues and are secreted into the blood and excreted in urine [[2]]. There are seven types of MPS that are categorized based on the lysosomal enzyme that is deficient (Table 1). Treatment options for MPSs may include ERT or hematopoietic stem cell therapy (HSCT) and other options such as gene therapy, genome editing, and PCs are being studied [[3]]. In this section, we will summarize the in vitro and in vivo studies reporting the identification and characterization of PC for MPS.
Table 1. Classification of the mucopolysaccharidoses (MPS).
|
Disorder |
Gene |
Enzyme Deficiency |
OMIM |
|
MPS I (Hurler, Hurler–Scheie, and Scheie syndrome) |
IDUA |
alpha-l-iduronidase |
607014 |
|
MPS II (Hunter syndrome) |
IDS |
Iduronate-2-sulfatase |
309900 |
|
MPS IIIA (Sanfilippo syndrome) |
SGSH |
Heparan-N-sulfatase |
252900 |
|
MPS IIIB (Sanfilippo syndrome) |
NAGLU |
N-acetylglucosaminidase |
252920 |
|
MPS IIIC (Sanfilippo syndrome) |
HGSNAT |
Acetyl CoA glucosamine N-acetyltransferase |
252930 |
|
MPS IIID (Sanfilippo syndrome) |
GNS |
N-acetyl-glucosamine-6-sulfatase |
252940 |
|
MPS IVA (Morquio A syndrome) |
GALNS |
N-acetylgalactosamine-6-sulfate sulfatase |
253000 |
|
MPS IVB (Morquio B syndrome) |
GLB1 |
β-galactosidase |
253010 |
|
MPS VI (Maroteaux–Lamy syndrome) |
ARSB |
Arylsulfatase B |
253200 |
|
MPS VII (Sly syndrome) |
GUSB |
β-glucuronidase |
253220 |
|
MPS IX |
HYAL1 |
Hyaluronidase |
601492 |
Mucopolysaccharidosis Type II
Mucopolysaccharidosis type II (MPS II, Hunter syndrome, OMIM 309900) is an X-linked disease caused by the deficiency of iduronate-2-sulafatase (IDS). This enzyme deficiency leads to the progressive lysosomal accumulation of the HS and DS, presenting systemic manifestations such as skeletal deformities, mental retardation, valvular heart disease, hepatosplenomegaly, and skin abnormalities [[4]].
Currently, ERT with recombinant human IDS (idursulfase and idursulfase-beta) is the standard treatment for MPS II patients. These ERTs have shown to be safe therapies for MPS II patients, and are effective in relation to functional capacity, urinary GAGs, and liver and spleen volume for attenuate and severe phenotypes. Nevertheless, limited effects have been observed in growth and cognitive disease [[3],[5]]. HSCT has not been recommended for MPS II patients due to the lack of CNS improvement of this treatment strategy [[4]].
Recently, a PC was described as a potential treatment alternative for MPS II patients [[4]]. In this study, the authors described the use of Δ-unsaturated 2-sulfouronic acid-N-sulfoglucosamine (D2S0, Figure 1A), which is a sulfated disaccharide derived from heparin. D2S0 has a similar structure to 2-O-sulfated iduronic acid-linked N-sulfated glucosamine, which is a natural substrate of IDS. The results showed that D2S0 has an IC50 of 30.1 µM and competes with the 4-methylumbelliferyl IDS artificial substrate, suggesting that this compound interacts with the active cavity of IDS. Recombinant human IDS (rhIDS) activity was significantly reduced (by about 25%) after incubation at 67 °C during 1 h; while the incubation with D2S0 increased, by about 5%, the thermal stability of rhIDS at this temperature, and in a dose-dependent manner. In MPS II fibroblasts, carrying the p.231L mutation, the treatment with 10 µM D2S0 allowed a 1.97-fold increase in IDS activity. The analysis of pathological iduronic acid from non-reducing end of GAGs did not show a significant change after 4 days post-treatment with D2S0; while a significant reduction in Alcian blue-positive granules (60%) was observed after 8 days post-treatment with D2S0. These results show the therapeutic potential of D2S0 and suggest that long D2S0 treatment could be necessary to reduce GAGs accumulation. In addition, the treatment of HEK293T cells expressing IDS mutants p.N63D, p.L67P, p.A85T, p.R88H, p.Y108S, and p.P231L with 10 µM D2S0 led to an increase in IDS activity between 1.6- and 39.6-fold, while no effect was observed in cells expressing p.L314P IDS. On the other hand, the p.A85T mutation showed a 7.7-fold reduction in IDS activity at 10 µM; at 0.1 µM a 1.7-fold increase was observed. In this sense, these results showed that D2S0 may function as a PC for IDS in a mutation-dependent manner. Nevertheless, enzyme activity values after treatment with D2S0 were between 2.0% and 7.0% of those observed for wild-type IDS, suggesting that further drug optimization through medicinal chemistry could be necessary to increase the efficacy of this molecule. However, this strategy could represent an interesting alternative for MPS II since about 41% of mutations affecting the IDS gene are missense. In addition, about 90% of these missense mutations are involved in amino acids that are buried within the fold of IDS, and it is expected that these mutations can induce local destabilization and misfolding, leading to premature degradation[[6]].
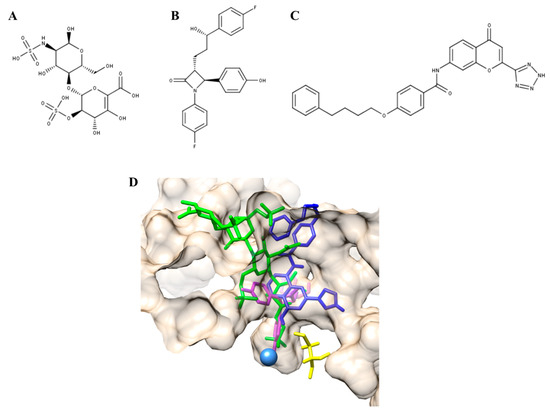
Figure 1. Pharmacological chaperones (PCs) for MPS II and MPS IVA. (A) Δ-unsaturated 2-sulfouronic acid-N-sulfoglucosamine (D2S0), (B) ezetimibe, and (C) pranlukast have been described as PC for (A) MPS II and (B,C) MPS IVA. (D) PC for MPS IVA were predicted to stablish similar interactions to those predicted for the natural and artificial N-acetylgalactosamine-6-sulfate sulfatase (GALNS) substrates. Green, KS; magenta, ezetimibe; blue, pranlukast. The catalytic residue (C79) and calcium ion are colored in yellow and light blue, respectively.
Mucopolysaccharidosis Type IVA
Mucopolysaccharidosis type IVA (MPS IVA, Morquio A syndrome, OMIM 252300) is an LSD caused by mutations in the gene encoding for the lysosomal enzyme N-acetylgalactosamine-6-sulfate sulfatase (GALNS) [[7]]. This enzyme deficiency leads to the lysosomal accumulation of the KS and chondroitin-6-sulfate (C6S) [[7],[8]]. Clinically, MPS IVA patients are characterized by short stature, corneal clouding, hypoplasia of the odontoid process, pectus carinatum, valvular heart disease, mild hepatomegaly, laxity of joints, kyphoscoliosis, and genu valgum without CNS impairment [[7],[8],[9]]. Treatment options for MPS IVA patients include non-steroidal anti-inflammatory drugs, antibiotics, oxygen supplementation, surgical procedures to correct orthopedic deformities and tracheal obstructions, ERT, and HSCT [[7],[8],[10]]. ERT has showed slight improvement in the 6-min walk test and the reduction of urinary KS [[11]], as well as an improvement in the maximal voluntary ventilation and performance of daily life activities [[12]]. Unfortunately, ERT has limited effects in correcting the abnormal growth and the skeletal, corneal, and cardiac abnormalities, due to tissue avascularity and short half-life of the enzyme [[13][14][15]]. On the other hand, although HSCT treatment showed long-term and normal enzyme activities, increase in lumbar bone mineral density, improvement in ambulatory movement and remission of the narrow airway [[16][17][18]], this is a high-risk procedure with many possible complications and high mortality. In this sense, it is necessary to explore new therapeutic strategies for MPS IVA to improve and expand the treatment options for MPS IVA patients.
Recently, the identification and characterization of the first PCs for MPS IVA was described [[19]]. Based on results from a modeled GALNS and computational dockings for natural and artificial substrates, a virtual screening strategy against 11,421 compounds tested in humans (ZINC In Man, a special subset of ZINC [[20]]), was designed to identify a set of compounds that bind to the active cavity of the enzyme. Ezetimibe and pranlukast (Figure 1B,C) were selected among the top 20 interacting compounds and were predicted to establish similar interactions to those modeled for KS and C6S, and the artificial GALNS substrate [[21],[19]] (Figure 1D). Ezetimibe is an approved drug that blocks intestinal cholesterol absorption by selectively inhibiting Niemann-Pick C1-like 1 protein and is indicated for treatment of disorders with elevated cholesterol levels [[21]]. Pranlukast is an orally administered, selective, and competitive cysteinyl leukotriene type 1 receptor antagonist, indicated for the treatment of bronchial asthma in pediatric and adult patients [[22]]. In terms of safety, both drugs are well tolerated and have adverse event profiles similar to placebo [[21],[22]].
In vitro evaluation showed that ezetimibe and pranlukast bind to the active cavity of the enzyme since they competed with the 4-methylumbelliferyl substrate (between 40% and 50% inhibition) and increased the thermal stability of the enzyme (5 °C). It was also observed that both compounds significantly increase the activity of human recombinant GALNS (hrGALNS) produced in bacteria (Escherichia coli), yeasts (Pichia pastoris), and mammalian cells (HEK293), with activities between 1.3- and 5.0-fold higher than in control cultures. This is an important observation, since protein folding represents a bottleneck in the production of recombinant proteins, reducing not only the protein’s biological activity, but also triggering cellular stress, which results in low production yields [[23][24]]. In this sense, these results show a potential application of ezetimibe and pranlukast to improve production yield and activity of hrGALNS, which may help to reduce immunogenicity of ERT, as the amount of protein used in ERT can be reduced [[25]].
Finally, both compounds were assayed in MPS IVA fibroblasts carrying the p.R61W, p.R94C, p.F285del, p.A393S, and p.W405_T406del mutations. In the case of ezetimibe, GALNS activity was increased in all treated cells between 1.4- and 2.5-fold compared to untreated cells, reaching, in some cases, similar levels to those observed in wild-type cells. On the other hand, pranlukast increased GALNS activity fibroblasts with p.R61W/p.W405_T406del and p.A393S mutations, albeit less effectively than ezetimibe. The results showed that the lower the concentration of the PC was, the greater the increase in the activity was, with the best results observed at 0.001 µM for both PCs. In addition, treatment of the MPS IVA fibroblasts with these PCs showed an increase in GALNS protein, reduction of lysosomal mass, and normalization of the autophagy markers LC3B and p62. Of note, it was observed that the combination therapy of hrGALNS with ezetimibe or pranlukast additively reduced lysosomal mass in patient-derived fibroblasts. In this sense, this study showed that ezetimibe and pranlukast are PCs that have the potential for use as a monotherapy for MPS IVA patients and could be also considered in the design of a combination therapy with ERT, which may improve the safety and efficacy of ERT for MPS IVA patients [[19]].
Mucopolysaccharidosis Type IVB
Deficiency of β-galactosidase (GLB1) causes two clinically distinct phenotypes: GM1-gangliosidosis (OMIM 230500) and mucopolysaccharidosis type IVB (MPS IVB, Morquio B syndrome, OMIM 253010). GLB1 deficiency leads to the impairment of the metabolism of GM1-gangliosides, glycoproteins, oligosaccharides, and KS, which accumulate in tissues and urine from patients with these clinical phenotypes [[26][27]]. GM1 gangliosidosis includes phenotypes that range from severe to mild, with a progressive CNS dysfunction that may include spasticity, deafness, blindness, decerebrate rigidity, insidious plateauing of motor and cognitive development followed by slow regression, gait disturbance, and cardiomyopathy. In some cases, skeletal involvement could be present including short stature, kyphosis, and scoliosis [[28]]. MPS IVB is characterized by generalized skeletal dysplasia including short-truck dwarfism, genu valgus, kyphosis, and short neck; as well as extra-skeletal manifestations such as corneal deposits, hepatomegaly, small teeth, and cardiac valvular lesions [[27]]. Currently, 185 mutations have been reported in GLB1, and about 70% of them are missense mutations. No specific treatment is available for these diseases and the patients are usually treated through symptomatic therapies [[26],[27]]. In contrast to MPS II and MPS IVA, several PCs have been described for β-galactosidase deficiencies (Figures 2–4 and Table 2). Although most of these studies have been performed for GM1-gangliosidosis-associated mutations, the results could be extrapolated to MPS IVB since the genetic defect affects the same enzyme.
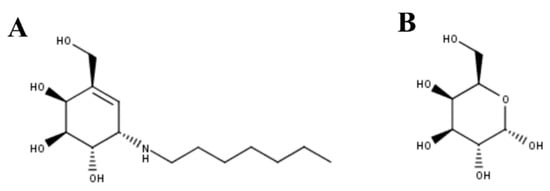
Figure 2. (A) N-octyl-4-epi-β-valienamine (NOEV) and (B) galactose were the first PCs described for GLB1 deficiency.
NOEV was the first PC described for GLB1 deficiency [[29]]. NOEV (Figure 2A) has an IC50 of 0.2 µM against human GLB1 and increased the enzyme activity between 1.2- and 5.1-fold in mouse fibroblasts expressing GLB1 carrying GM1-gangliosidosis (p.R201C and p.R201H/p.R457Q) or MPS IVB (p.W273L/p.Y83H) mutations. Similar results were observed in human fibroblasts from GM1-gangliosidosis patients. In vivo evaluation of NOEV was performed in a mouse expressing the human p.R201C-mutant GLB1 but lacking the endogenous mouse β-galactosidase. Oral administration of NOEV (1 mM ad libitum for 1 week, which corresponds approximately to 1.4 mg per day) led to a significant increase of GLB1 activity in cerebrum, cerebellum, heart, lung, liver, spleen, kidney, muscle, and plasma; with heart, lung, spleen, and muscle showing the greatest improvements. As observed by an immunostaining analysis of the brain, GM1 and GA1 storage was reduced in the frontotemporal cerebral cortex and brainstem, which is a surprising result due to the short term of the treatment. In fact, mass biochemical analysis of brains from treated mice (i.e., lipids analysis by TLC) did not show reduction in GM1 and GA1. In this sense, although the results showed the potential of NOEV as a PC for GLB1 deficiencies, long-term studies should be performed to establish the optimal dose to evaluate the effect of this PC on substrate reduction, and to observe the potential adverse effects.
Galactose was then evaluated as potential chemical chaperone for GLB1 (Figure 2B) [[30]]. Galactose competitively bound to the active cavity of β-galactosidase, since a significant reduction in enzyme activity (92%) was observed in wild-type human fibroblasts treated with 200 mM galactose. Treatment of GM1-gangliosidosis patient fibroblasts with 200 mM galactose showed a 2.5-fold increase in enzyme activity in cells carrying the p.T329A/p.R442Q mutations; while no effect was observed in fibroblasts carrying the p.S54N/p.R59C and p.R201H/p.G579D mutations. Similarly, the addition of 200 mM galactose to COS-1 cells expressing a p.R442Q mutated β-galactosidase showed a 1.7-fold increase in enzyme activity. These results show the potential of galactose as a PC for GLB1 deficiency.
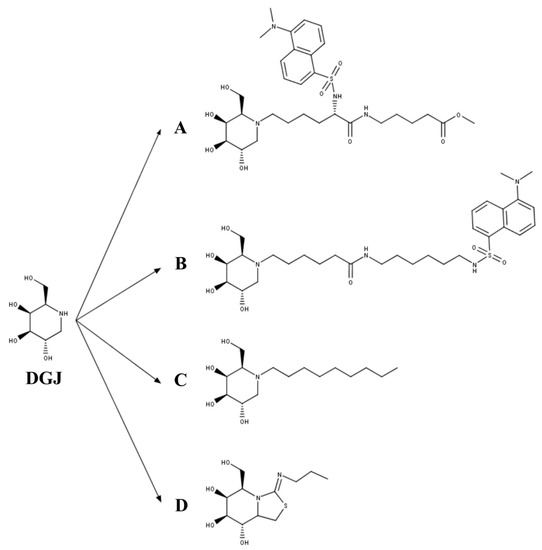
Figure 3. The 1-deoxygalactonojirimycin (DGJ)-derived PCs for GLB1 deficiency. (A) Methyl 6-{[N2-(dansyl)-N6-(1,5-dideoxy-d-galactitol-1,5-diyl)-L-lysyl]amino} hexanoate (DLHex-DGJ); (B) N-(dansylamino) hexylaminocarbonylpentyl-1,5-dideoxy-1,5-imino-d-galactitol; (C) N-nonyl-deoxygalactonojirimycin (NN-DGJ); and (D) 5N,6S-(N-butyliminomethylidene)-6-thio-1-deoxygalactonojirimycin (6S-NBI-DGJ).
A third set of PCs for GLB1 have been derived from DGJ (Figure 3), a well-known competitive inhibitor of lysosomal glycosidases that is approved for the treatment of Fabry disease [[31]]. Fantur et al. [[32]] evaluated the compound methyl 6-{[N2-(dansyl)-N6-(1,5-dideoxy-d-galactitol-1,5-diyl)-L-lysyl]amino} hexanoate (DLHex-DGJ) as a PC for GLB1 (Figure 3A). It was expected that the chemical modifications done on the DGJ core to produce DLHex-DGJ increased the lipophilicity and the inhibition activity compared to DGJ. DLHex-DGJ showed a Ki of 0.6 µM and an IC50 of 6 µM; it was 10-fold more effective than DGJ, resulting in approximately 94% inhibition of a human wild-type GLB1. The treatment of GM1-gangliosidosis and MPS IVB patient fibroblasts with 20 to 500 µM DLHex-DGJ showed a 1.3- to 12.5-fold increase in GLB1 activity depending on the mutation, reaching values between 0.4% and 57.2% of those from wild-type fibroblasts (Table 2). These results showed that although DLHex-DGJ increased the activity of all tested mutations, this increment could not be enough to have clinical impact on the disease phenotype. Of note, the highest increase in enzyme activity was observed on fibroblasts carrying mutations at p.R201 (p.R201C and p.R201H), which were associated with production of misfolded and unstable precursor proteins rapidly degraded by ERAD. In these mutations, treatment with DLHex-DGJ enhanced the amount of mature enzyme and restored the intracellular trafficking of the enzyme. Similarly, Fröhlich et al. [[33]] evaluated the chaperone capacity of the N-(dansylamino)hexylaminocarbonylpentyl-1,5-dideoxy-1,5-imino-d-galactitol (Figure 3B). This compound showed a Ki of 0.7 µM in human GLB1, and a greater chaperone effect than DLHex-DGJ, since significant increase in enzyme activity was observed even at 1 µM (approximately 2-fold increase) reaching up to a 10-fold increase after 500 µM treatment of human fibroblasts with the p.R201C mutation. Nevertheless, the results showed that N-(dansylamino) hexylaminocarbonylpentyl-1,5-dideoxy-1,5-imino-d-galactitol was less effective than NOEV. A third DGJ-derived PC evaluated for GLB1 deficiency was N-nonyl-deoxygalactonojirimycin (NN-DGJ, Figure 3C) [[34]]. This compound is a competitive inhibitor of human GLB1 and has an IC50 of 0.04 µM at pH 6.5 and of 0.120 µM at pH 4.3, thus suggesting a lower affinity at the lysosome than in the ER. NN-DGJ also increased the thermostability of wild-type human GLB1 between 3- and 4-fold, confirming the direct interaction of NN-DGJ with the enzyme and the chaperone activity of NN-DGJ. Evaluation in GM1 fibroblasts carrying the mutations p.R148S/p.D332N showed that a significant increase in GLB1 activity was only observed with concentrations 15-fold greater than that of the IC50, reaching a 2- to 3-fold increase in activity with concentrations between 1.0 and 2.6 µM. Treatment of GM1 fibroblasts with 1.2 µM NN-DGJ restored the intracellular trafficking of the mutated enzyme, reducing the GLB1 present in the ER and increasing the amount of GLB1 in the lysosome. Evaluation on GM1 and MPS IVB human fibroblasts showed 5- and 7-fold increases in enzyme activity on fibroblasts with mutations p.R201H/IVS14-2A>G and p.R201H/p.W509C, respectively, favoring also a correct maturation of these mutated enzymes. However, other GM1 and MPS IVB fibroblasts were not responsive to the treatment with NN-DGJ. Finally, a bicyclic DGJ derivative was evaluated as a novel PC for GLB1 [[35]]. This derivative, namely 5N,6S-(N-butyliminomethylidene)-6-thio-1-deoxygalactonojirimycin (6S-NBI-DGJ, Figure 3D), inhibits wild-type human GLB1 with an IC50 of 32 µM, and significantly increases the thermostability of the enzyme. Treatment of GM1 patient fibroblasts with 20 and 80 µM 6S-NBI-DGJ showed a significant improvement of GLB1 activity (between 2- and 3-fold) for those fibroblasts carrying the mutations p.I51T, p.I51T/p.Y316C, p.I51T/p.R457Q, p.G190D, p.R201C, p.G438E, and p.R457Q; no response was observed for p.R59H mutation. The chaperone activity of 6S-NBI-DGJ was also observed on 24 (27%) out of 88 mutated GLB1 enzymes expressed in COS7 cells. This compound significantly reduced the accumulation of GM1 ganglioside in patient fibroblasts, as well as the levels of the autophagy biomarkers p62 and LC3-II. Oral treatment of a p.R201C GM1 gangliosidosis mouse model led to a significant increase in GLB1 activity in heart, liver, kidney, and brain, and to the reduction of GM1 gangliosides and autophagy biomarker (p62 and LC3-II) levels in brain samples. Overall, DGJ-derived compounds have a potential as PC for GLB1 deficiencies, since they increase the enzyme activity of an important number of mutations. Nevertheless, most of the studied MPS IVB mutations seem to be not responsive to these DGJ-derived PC, and a more detailed study of MPS IVB mutations is still required.
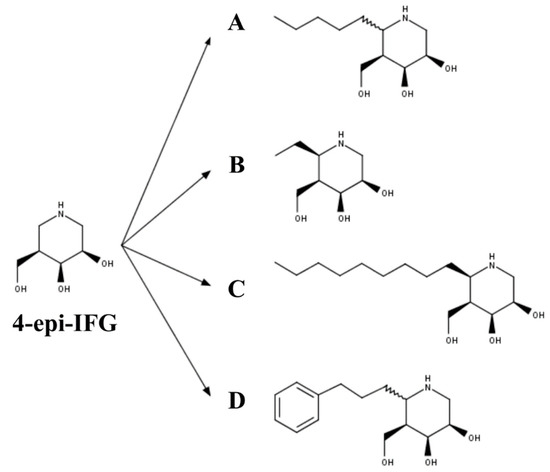
Figure 4. The 4-epi-isofagomine (4-epi-IFG) derived PC for GLB1 deficiency: (A) (5aS)- and (5aR)-5a-C-pentyl-4-epi-IFG; (B) 5a-C-methyl-4-epi-IFG; (C) 5a-C-nonyl-4-epi-IFG; and (D) 5a-C-2-phenylethyl-4-epi-IFG.
Finally, 4-epi-IFG-derived compounds have also been studied as PCs for GLB1 deficiencies (Figure 4). In the first case, the compounds (5aS)- and (5aR)-5a-C-pentyl-4-epi-IFG were evaluated (Figure 4A) [[35]]. Inhibition studies using a wild-type human GLB1 showed that (5aR)-5a-C-pentyl-4-epi-IFG was about 1600-fold more active than the (5aS)-based compound, with an IC50 of 0.008 µM at pH 7.3 and an IC50 of 0.180 µM at pH 4.3, suggesting a lower affinity of this compound for GLB1 at the lysosome pH. As observed with other PC for GLB1, (5aR)-5a-C-pentyl-4-epi-IFG increased the thermostability of the human wild-type enzyme in a dose-dependent manner. The effect on mutated GLB1 was evaluated in 23 human fibroblasts carrying different missense mutations (Table 2), two of which were associated with an MPS IVB phenotype (p.W273L/p.R482H and p.W273L/p.W509C). An increase in the enzyme activity was observed in 56% of the evaluated mutations, which ranged from 1.5- to 35-fold. Specifically, MPS IVB fibroblasts showed a 1.5-fold increase in the GLB1 activity. Recently, 4-epi-IFG-derived compounds carrying methyl, nonyl, and 2-phenylethyl groups at C-5a (Figure 4B–D) were evaluated as potential PCs for human GLB1 [[35]]. Whereas the 2-phenylethyl- and nonyl-derivates showed IC50 similar to or less than that of the previously studied (5aR)-5a-C-pentyl-derived compound, the methyl-derived compound showed a significantly (25-fold) higher IC50 than the (5aR)-5a-C-pentyl-derived compound. These results correlated with the enhancement of the enzyme activity in GM1 gangliosidosis patient fibroblasts, with nonyl-derivate showing the most efficient chaperone activity and requiring a 50-fold lower concentration than the (5aR)-5a-C-pentyl- and 2-phenylethyl-derivates to reach the same enzyme activity increase. Nevertheless, nonyl-derivate showed a significantly higher cytotoxicity than the other 4-epi-IFG-derived compounds evaluated, which confirmed the potential of (5aR)-5a-C-pentyl-4-epi-IFG as a PC for GLB1 deficiency. In fact, the treatment of GM1 gangliosidosis patient fibroblasts with (5aR)-5a-C-pentyl-4-epi-IFG restored the maturation of the mutated enzyme and led to the reduction of 31% and 36% of KS and Hex3HexNAc2 oligosaccharide, respectively.
Table 2. Summary of the effect on enzyme activity of pharmacological chaperones for GLB1 deficiencies.
|
Pharmacological Chaperone |
IC50/Ki (µM) |
GLB1 Mutations |
Maximum Activity Enhancement (Fold Change) |
Reference |
|
NOEV |
IC50: 0.2 |
Patient Fibroblasts p.R201C p.R201H p.R457Q p.W273L * p.Y83H * |
5.1 4.5 2.4 2.2 2.0 |
[102] |
|
Galactose |
N.D. |
Patient Fibroblasts p.S54N/p.R59C p.R201/p.G579D p.T329A/p.R442Q |
1.0 1.4 2.6 |
[103] |
|
DLHex-DGJ |
IC50: 6.0 Ki: 0.6 |
Patient Fibroblasts p.I181K p.R201C p.R201H/p.R457X p.R201H/p.S149F p.R201H/p.H281Y p.R208C/p.W161X p.C230R p.Y270D p.W273L p.A301V p.Y333H p.G438E p.P549L |
2.1 9.4 11.1 12.3 12.5 2.5 9.0 1.7 1.3 1.3 1.8 2.3 1.4 |
[104] |
|
N-(dansylamino)hexylaminocarbonylpentyl-DGJ |
Ki: 0.7 |
Patient Fibroblasts p.R201C |
~7.0 |
[[33]] |
|
NN-DGJ |
IC50: 0.12 |
Patient Fibroblasts p.R351X/p.R351X p.R148S/p.D332N p.R148S/p.R482H p.R201H/IVS14-2A>G p.R201H/p.W509C p.W273L/p.R482H * p.G438E/p.G438E * |
1.4 4.1 4.9 7.3 0.9 1.0 1.0 |
|
|
6S-NBI-DGJ |
IC50: 32 |
Patient Fibroblasts p.I51T p.I51T/p.Y316C p.I51T/p.R457Q p.R59H p.G190D p.R201C p.G438E p.R457Q COS7 cells p.I51T p.R148T p.L155R p.G190D p.R201C p.R201H p.R208C p.D214Y p.V216A p.C230Y p.L264S p.N266S p.W273R p.D332N p.K346N p.S434L p.G438E p.Y444C p.R457Q p.R482H p.D491Y p.R590H p.E632G p.D640E |
2.6 5.5 4.9 1.1 4.1 4.9 2.7 3.0
1,9 1,8 1,6 1,5 1,9 2,5 1,8 1,4 1,7 2,3 1,9 2,0 1,9 2,2 1,4 2,2 2,2 2,8 2,4 1,5 1,7 2,0 1,6 1,7 |
[[35]] |
|
(5aR)-5a-C-pentyl-4-epi-IFG |
IC50: 0.008 |
Patient Fibroblasts 733 + 2T > C p.I51T p.R59H p.R59H p.C127Y p.R148S/p.S532G/p.L305F p.S191N/R351X p.R201C p.R201C p.R201C/p.H281Y p.R201H/c.247dup1 p.R201H/p.G76E p.R201H/ p.H281Y p.R208C/IVS10 + 1G > A p.Q255H/p.K578R p.H281Y/splicing p.R351Ter/p.R351X p.G438E p.R442Q/p.W92X p.R457Q p.P549L p.W273L/p.R482H * p.W273L/ p.W509C * |
1.1 1.6 1.4 1.1 1.1 1.0 11.0 15.0 5.4 18.0 3.5 6.7 4.7 4.4 20.0 35.0 1.0 2.0 1.5 7.3 1.3 1.5 1.5 |
[[35]] |
N.D.: not determined. * Mutations associated with an MPS IVB phenotype. GLB1 mutations were evaluated in patient fibroblasts or in COS7 cells overexpressing the mutated enzyme
This entry is adapted from the peer-reviewed paper 10.3390/ijms21010232
References
- Lorne Clarke; Carolyn Ellaway; Helen E. Foster; Roberto Giugliani; Cyril Goizet; Sarah Goring; Sara Hawley; Elaina Jurecki; Zaeem Khan; Christina Lampe; et al. Understanding the Early Presentation of Mucopolysaccharidoses Disorders. Journal of Inborn Errors of Metabolism and Screening 2018, 6, no, 10.1177/2326409818800346.
- Shaukat A. Khan; Hira Peracha; Diana Ballhausen; Alfred Wiesbauer; Marianne Rohrbach; Matthias Gautschi; Robert W. Mason; Roberto Giugliani; Yasuyuki Suzuki; Kenji E. Orii; et al. Epidemiology of mucopolysaccharidoses.. Molecular Genetics and Metabolism 2017, 121, 227-240, 10.1016/j.ymgme.2017.05.016.
- Kazuki Sawamoto; Molly Stapleton; Carlos J. Alméciga-Díaz; Angela J. Espejo-Mojica; Juan Camilo Losada; Diego A. Suarez; Shunji Tomatsu; Therapeutic Options for Mucopolysaccharidoses: Current and Emerging Treatments. Drugs 2019, 79, 1103-1134, 10.1007/s40265-019-01147-4.
- Tomatsu, S.; Kubasky, F.; Stapleton, M.; Suzuki, Y.; Orii, K.; Vairo, F.; Brusius-Facchin, A.C.; Leistner-Segal, S.; Graeff, M.; Fischinger, C.; et al. Mucopolysaccharidosis Type II: Clinical Features, Biochemistry, Diagnosis, Genetics, and Treatment. In Mucopolysaccharidoses Update (2 Volume Set); Tomatsu, S., Lavery, C., Giugliani, R., Harmatz, P., Scarpa, M., Węgrzyn, G., Orii, T., Eds.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2018; Volume I, pp. 165–209
- Hiroo Hoshina; Yohta Shimada; Takashi Higuchi; Hiroshi Kobayashi; Hiroyuki Ida; Toya Ohashi; Chaperone effect of sulfated disaccharide from heparin on mutant iduronate-2-sulfatase in mucopolysaccharidosis type II. Molecular Genetics and Metabolism 2018, 123, 118-122, 10.1016/j.ymgme.2017.12.428.
- Mykhaylo Demydchuk; Chris H. Hill; Aiwu Zhou; Gábor Bunkóczi; Penelope E. Stein; Denis Marchesan; Janet E. Deane; Randy J. Read; Insights into Hunter syndrome from the structure of iduronate-2-sulfatase. Nature Communications 2017, 8, 15786, 10.1038/ncomms15786.
- Sawamoto, K.; Alméciga-Díaz, C.J.; Mason, R.W.; Orii, T.; Tomatsu, S. Mucopolysaccharidosis Type IVA: Clinical Features, Biochemistry, Diagnosis, Genetics, and Treatment. In Mucopolysaccharidoses Update (2 Volume Set); Tomatsu, S., Lavery, C., Giugliani, R., Harmatz, P., Scarpa, M., Węgrzyn, G., Orii, T., Eds.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2018; Volume I, pp. 235–272
- Shaukat Khan; Carlos J. Alméciga-Díaz; Kazuki Sawamoto; William G. MacKenzie; Mary C. Theroux; Christian Pizarro; Robert W. Mason; Tadao Orii; Shunji Tomatsu; Mucopolysaccharidosis IVA and glycosaminoglycans.. Molecular Genetics and Metabolism 2016, 120, 78-95, 10.1016/j.ymgme.2016.11.007.
- S. Tomatsu; A. M. Montaño; H. Oikawa; Daniel J. Rowan; M. Smith; L. Barrera; Y. Chinen; M. M. Thacker; W. G. MacKenzie; Y. Suzuki; et al. Mucopolysaccharidosis Type IVA (Morquio A Disease): Clinical Review and Current Treatment: A Special Review. Current Pharmaceutical Biotechnology 2011, 12, 931-945, 10.2174/138920111795542615.
- Madeleine Taylor; Shaukat Khan; Molly Stapleton; Jianmin Wang; Jing Chen; Robert Wynn; Hiromasa Yabe; Yasutsugu Chinen; Jaap Jan Boelens; Robert W. Mason; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biology of Blood and Marrow Transplantation 2019, 25, e226-e246, 10.1016/j.bbmt.2019.02.012.
- Christian J. Hendriksz; Barbara Burton; Thomas R. Fleming; Paul Harmatz; Derralynn Hughes; Simon A. Jones; Shuan-Pei Lin; Eugen Mengel; Maurizio Scarpa; Vassili Valayannopoulos; et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study.. Journal of Inherited Metabolic Disease 2014, 37, 979-990, 10.1007/s10545-014-9715-6.
- Christian J. Hendriksz; Roberto Giugliani; Paul Harmatz; Eugen Mengel; Nathalie Guffon; Vassili Valayannopoulos; Rossella Parini; Derralynn Hughes; Gregory M. Pastores; Heather A. Lau; et al. Multi-domain impact of elosulfase alfa in Morquio A syndrome in the pivotal phase III trial. Molecular Genetics and Metabolism 2015, 114, 178-185, 10.1016/j.ymgme.2014.08.012.
- Tomatsu, S.; Sawamoto, K.; Shimada, T.; Bober, M.B.; Kubaski, F.; Yasuda, E.; Mason, R.W.; Khan, S.; Alméciga-Díaz, C.J.; Barrera, L.A.; et al. Enzyme replacement therapy for treating mucopolysaccharidosis type IVA (Morquio A syndrome): Effect and limitations. Expert Opin. Orphan Drugs 2015, 3, 1279–1290.
- Caitlin Doherty; Molly Stapleton; Matthew Piechnik; Robert W. Mason; William G. MacKenzie; Seiji Yamaguchi; Hironori Kobayashi; Yasuyuki Suzuki; Shunji Tomatsu; Effect of enzyme replacement therapy on the growth of patients with Morquio A. Journal of Human Genetics 2019, 64, 625-635, 10.1038/s10038-019-0604-6.
- Becky Schweighardt; Troy Tompkins; Kelly Lau; Lynne Jesaitis; Yulan Qi; Donald G. Musson; Pamela Farmer; Christine Haller; Adam J. Shaywitz; Ke Yang; et al. Immunogenicity of Elosulfase Alfa, an Enzyme Replacement Therapy in Patients With Morquio A Syndrome: Results From MOR-004, a Phase III Trial. Clinical Therapeutics 2015, 37, 1012-1021.e6, 10.1016/j.clinthera.2014.11.005.
- Yasutsugu Chinen; Takeshi Higa; Shunji Tomatsu; Yasuyuki Suzuki; Tadao Orii; Nobuyuki Hyakuna; Long-term therapeutic efficacy of allogenic bone marrow transplantation in a patient with mucopolysaccharidosis IVA. Molecular Genetics and Metabolism Reports 2014, 1, 31-41, 10.1016/j.ymgmr.2013.11.002.
- Shunji Tomatsu; Hiromasa Yabe; Akemi Tanaka; Yasutsugu Chinen; Shunichi Kato; Yasuyuki Suzuki; Tadao Orii; Hematopoietic stem cell transplantation for Morquio syndrome type A. Molecular Genetics and Metabolism 2016, 117, S114, 10.1016/j.ymgme.2015.12.464.
- Jianmin Wang; Zuo Luan; Hua Jiang; Jianpei Fang; Maoquan Qin; Vincent Lee; Jing Chen; Allogeneic Hematopoietic Stem Cell Transplantation in Thirty-Four Pediatric Cases of Mucopolysaccharidosis-A Ten-Year Report from the China Children Transplant Group.. Biology of Blood and Marrow Transplantation 2016, 22, 2104-2108, 10.1016/j.bbmt.2016.08.015.
- Carlos J. Alméciga-Diaz; Oscar A. Hidalgo; Sergio Olarte-Avellaneda; Alexander Rodriguez-Lopez; Esteban Guzman; Rafael Garzón; Luisa Natalia Pimentel-Vera; Maria Alejandra Puentes-Tellez; Andres Felipe Rojas-Rodriguez; Kirill Gorshkov; et al. Identification of Ezetimibe and Pranlukast as Pharmacological Chaperones for the Treatment of the Rare Disease Mucopolysaccharidosis Type IVA. Journal of Medicinal Chemistry 2019, 62, 6175-6189, 10.1021/acs.jmedchem.9b00428.
- Peter P Toth; Binh An P Phan; Thomas D Dayspring; Ezetimibe therapy: mechanism of action and clinical update. Vascular Health and Risk Management 2012, 8, 415-427, 10.2147/VHRM.S33664.
- Sergio Olarte-Avellaneda; Alexander Rodriguez-Lopez; Carlos Javier Alméciga-Díaz; Luis Alejandro Barrera; Computational analysis of human N-acetylgalactosamine-6-sulfate sulfatase enzyme: an update in genotype–phenotype correlation for Morquio A. Molecular Biology Reports 2014, 41, 7073-7088, 10.1007/s11033-014-3383-3.
- Susan J Keam; Katherine A Lyseng-Williamson; Karen L Goa; Pranlukast: a review of its use in the management of asthma.. Drugs 2003, 63, 991-1019, .
- Marizela Delic; Rebecca Göngrich; Diethard Mattanovich; Brigitte Gasser; Engineering of Protein Folding and Secretion—Strategies to Overcome Bottlenecks for Efficient Production of Recombinant Proteins. Antioxidants & Redox Signaling 2014, 21, 414-437, 10.1089/ars.2014.5844.
- Brigitte Gasser; Roland Prielhofer; Hans Marx; Michael Maurer; Justyna Nocon; Matthias Steiger; Verena Puxbaum; Michael Sauer; Diethard Mattanovich; Pichia pastoris: protein production host and model organism for biomedical research. Future Microbiology 2013, 8, 191-208, 10.2217/fmb.12.133.
- Coen Maas; Suzanne Hermeling; Barend Bouma; Wim Jiskoot; Martijn F. B. G. Gebbink; A Role for Protein Misfolding in Immunogenicity of Biopharmaceuticals. Journal of Biological Chemistry 2006, 282, 2229-2236, 10.1074/jbc.m605984200.
- Debra S Regier; Richard L Proia; Alessandra D'azzo; Cynthia J Tifft; The GM1 and GM2 Gangliosidoses: Natural History and Progress toward Therapy.. Pediatr Endocrinol Rev 2016, 13, , .
- Higaki, K.; Ninomiya, H. Mucopolysaccharidosis Type IVB: Clinical Features, Biochemistry, Diagnosis, Genetics, and Treatment. In Mucopolysaccharidoses Update (2 Volume Set); Tomatsu, S., Lavery, C., Giugliani, R., Harmatz, P., Scarpa, M., Węgrzyn, G., Orii, T., Eds.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2018; Volume I, pp. 273–283.
- Regier, D.S.; Tifft, C.J. GLB1-Related Disorders. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; GeneReviews®: Seattle, WA, USA, 1993
- Junichiro Matsuda; Osamu Suzuki; Akihiro Oshima; Yoshie Yamamoto; Akira Noguchi; Kazuhiro Takimoto; Masayuki Itoh; Yuji Matsuzaki; Yosuke Yasuda; Seiichiro Ogawa; et al. Chemical chaperone therapy for brain pathology in GM1-gangliosidosis. Proceedings of the National Academy of Sciences 2003, 100, 15912-15917, 10.1073/pnas.2536657100.
- Anna Caciotti; Maria Alice Donati; Alessandra D'azzo; Rosa Salvioli; Renzo Guerrini; Enrico Zammarchi; Amelia Morrone; The potential action of galactose as a “chemical chaperone”: Increase of beta galactosidase activity in fibroblasts from an adult GM1-gangliosidosis patient. European Journal of Paediatric Neurology 2009, 13, 160-164, 10.1016/j.ejpn.2008.03.004.
- Derralynn A Hughes; Kathleen Nicholls; Suma P Shankar; Gere Sunder-Plassmann; David Koeller; Khan Nedd; Gerard Vockley; Takashi Hamazaki; Robin Lachmann; Toya Ohashi; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. Journal of Medical Genetics 2016, 54, 288-296, 10.1136/jmedgenet-2016-104178.
- Katrin Fantur; Doris Hofer; Georg Schitter; Andreas J. Steiner; Bettina M. Pabst; Tanja M. Wrodnigg; Arnold E. Stütz; Eduard Paschke; DLHex-DGJ, a novel derivative of 1-deoxygalactonojirimycin with pharmacological chaperone activity in human GM1-gangliosidosis fibroblasts. Molecular Genetics and Metabolism 2010, 100, 262-268, 10.1016/j.ymgme.2010.03.019.
- Richard F.G. Fröhlich; Katrin Fantur; Richard H. Furneaux; Eduard Paschke; Arnold E. Stütz; Jacqueline Wicki; Stephen G. Withers; Tanja M. Wrodnigg; A fluorescent probe for GM1 gangliosidosis related β-galactosidase: N-(Dansylamino)hexylaminocarbonylpentyl-1,5-dideoxy-1,5-imino-d-galactitol. Bioorganic & Medicinal Chemistry Letters 2011, 21, 6872-6875, 10.1016/j.bmcl.2011.09.012.
- Brigitte A. Rigat; Michael B. Tropak; Justin Buttner; Ellen Crushell; Daphne Benedict; John W. Callahan; Uglas R. Martin; Don J. Mahuran; Evaluation of N-nonyl-deoxygalactonojirimycin as a pharmacological chaperone for human GM1 gangliosidosis leads to identification of a feline model suitable for testing enzyme enhancement therapy.. Molecular Genetics and Metabolism 2012, 107, 203-212, 10.1016/j.ymgme.2012.06.007.
- Tomoko Takai; Katsumi Higaki; Matilde Aguilar-Moncayo; Teresa Mena-Barragán; Yuki Hirano; Kei Yura; Liang Yu; Haruaki Ninomiya; María Isabel Garcia Moreno; Yasubumi Sakakibara; et al. A Bicyclic 1-Deoxygalactonojirimycin Derivative as a Novel Pharmacological Chaperone for GM1 Gangliosidosis. Molecular Therapy 2013, 21, 526-532, 10.1038/mt.2012.263.