Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, General & Internal
Renal disorders are very prevalent and associated with significant morbidity and mortality. Among diverse pathogenic mechanisms, the dysregulation of immune and inflammatory responses plays an essential role in such disorders. Consequently, the discovery of Annexin A1, as a glucocorticoid-inducible anti-inflammatory protein, has fueled investigation of its role in kidney pathologies. Thus, its role has been examined in diverse renal pathologies, including acute kidney injury, diabetic nephropathy, immune-mediated nephropathy, drug-induced kidney injury, kidney stone formation, and renal cancer.
- annexins
- kidney
- acute kidney injury
- kidney stone
1. Introduction
Annexins were discovered over 40 years ago and constitute a superfamily of more than a thousand proteins, in major eukaryotic phyla, with complex and diverse functions including calcium metabolism, intracellular signaling, subcellular transport, membrane repair, cell adhesion, cell death/survival, growth and differentiation, among others. Structurally, the C-terminus of annexins is comprised of four homologous repeats of about 70 amino acids which contain the endonexin sequence (GxGT-38 residues) that serves as a (type 2) calcium binding site with high affinity for calcium and phospholipids. The variable N-terminus harbors sites for post-translational modifications and protein-protein interactions [1][2][3]. Although substantial homologies exist among the superfamily of annexins, variations in amino acid sequences among different groups of eukaryotes translate into structural characteristics and biochemical features causing eukaryotic group-specific functional differences. The current nomenclature for annexins takes into consideration the evolutionary divisions of the eukaryotes and refers to those of the vertebrates, including human, as family A with the 13 annexins commonly found in vertebrates named as Annexin A1 to A13 (also known as ANXA1-ANXA13) [1][2][3][4][5][6]. Human annexin-A7 (synexin) was the first to be isolated and purified and human Annexin A1 (lipocortin) and -A2 (calpactin) were the first to be cloned [1][2].
Annexin A1, the product of ANXA1 gene, is a 37 kDa protein that was initially discovered (in the 1980s) as a dexamethasone (DEXA)-inducible protein (e.g., in renal cells). It has also been referred to as lipocortin-1, renocortin, and macrocortin [5][6]. It is now known that Annexin A1 is expressed in variety of tissues (e.g., heart, brain, blood vessels) and that immune cells (e.g., leukocytes) display high expression levels. At the cellular level, Annexin A1 is expressed in the nucleus, cytoplasm, and cell membrane, consistent with its multifaceted functions (e.g., proliferation, apoptosis, survival, differentiation and migration, anti-inflammatory, etc.) [7]. The ability of glucocorticoids to regulate mRNA transcription of Annexin A1 has been shown in in vitro (e.g., using a human epithelial cell line A549), ex vivo (e.g., alveolar macrophages of patients with inflammatory lung disease), and in vivo (e.g., rat brain) studies [8]. Importantly, aside from transcription, DEXA also causes the translocation of Annexin A1 to the cell membrane and its secretion, processes likely mediated through serine-27 phosphorylation via mitogen-activated protein kinase- (MAPK), phosphatidylinositol-3-kinase- (PI3K), and protein kinase C-dependent signaling pathways (e.g., in a human folliculostellate cell line) [9]. Importantly, cell-specific molecular events culminating in the secretion of Annexin A1 are of major relevance and implication for the binding and attachment of leukocytes onto endothelial surfaces [8]. For example, ATP binding cassette transporter system is believed to regulate Annexin A1 secretion in macrophages while in neutrophils cytoplasmic Annexin A1 is stored in gelatinase granules and is released in response to chemoattractants or adhesion to endothelial cells monolayers [10]. Aside from its potent inhibitory effects on neutrophil trafficking, several other major effects of Annexin A1 on leukocytes have been described, including the (a) release of Annexin A1 from apoptotic neutrophils promoting macrophage phagocytosis and removal of apoptotic cells, (b) Annexin A1 secretion by macrophages (e.g., in response to glucocorticoids), acting in autocrine or paracrine fashion, increasing entrapment of apoptotic neutrophils, and (c) exogenous administration of Annexin A1 increasing human neutrophil apoptosis mediated, in part, via dephosphorylation of pro-apoptotic protein- Bad [10].
One of the early discoveries which subsequently led to further unraveling of the anti-inflammatory effects of Annexin A1 is its inhibition of cytosolic phospholipase A2 (PLA2) and subsequent blockade of arachidonic acid release leading to the inhibition of generation of eicosanoids (e.g., prostaglandins such as PGE2) [5][11][12][13][14][15][16]. Further, as indicated above, Annexin A1 is also transported to the cell membrane and externalized or secreted into the extracellular fluid which, in turn, exerts its effects (e.g., anti-inflammatory) [7]. Importantly, the myriad of effects attributed to Annexin A1, as well as its N-terminal-derived peptides (i.e., Ac2-26, Ac2-12, and Ac2-6), are believed to be mediated via binding to G-protein coupled receptors, e.g., formyl peptide receptors 1 and 2 (Fpr1, Fpr2), and/or its ability to bind to cell membrane phospholipids [8][17][18]. Fpr2 is the main receptor for anti-inflammatory effects of Annexin A1. It is also known as the ALX as it also serves as the receptor for the anti-inflammatory molecule lipoxin A4 (i.e., Fpr2/ALX) [10]. Accordingly, extracellular Annexin A1, in the presence of ≥1 mM Ca2+, undergoes conformational change that leads to exposure of the N-terminal region and binding thereby facilitating its interaction with Fpr2/ALX. Annexin A1 can function in an autocrine, paracrine, and juxtacrine (i.e., involving cell-cell contact) fashion to activate Fpr2/ALX signaling; the latter mechanism is suggested as the most plausible mechanism for the actions of Annexin A1 in inflammatory conditions [10]. The Annexin A1-induced Fpr2/ALX signaling involves phosphorylation of extracellular-regulated kinase (ERK) 1/2 that is associated with rapid elevation in intracellular calcium concentration. Indeed, blockade of Fpr2/ALX signaling, using a monoclonal antibody, inhibited Annexin A1-induced human neutrophil transmigration and adhesion to endothelial-cell monolayers under flow [19].
The early recognition of the role of Annexin A1 as an anti-inflammatory protein has been followed by the unraveling of a myriad of its effects, with therapeutic potential, in various pathologies, including those involving the central nervous system, cardiovascular system, the respiratory system, and the gastrointestinal tract, among others. These aspects have been the focus of previous reviews [8][20][21][22][23][24][25]. Thus, herein is intended to primarily describe the progress made in unraveling the role of Annexin A1 in the kidney, in health and disease, which should help identify venues of further research in this field.
2. Annexins in the Kidney
Early immunohistochemical studies, of normal rat kidney, showed a marked presence of Annexin A1 in epithelia of Bowman’s capsule, the macula densa, and medullary/papillary collecting ducts, but mild staining in the thick ascending limb of the loop of Henle, while other segments were largely devoid of staining [4][26] (Figure 1). Others reported four annexins (with apparent molecular masses of about 32, 35, 45 and 67 kDa and labelled as I, II, V, and VI) in isolated rat renal glomeruli and glomerular mesangial cells [27]. More recently, prominent Annexin A1 expression has been demonstrated for renal interstitial fibroblasts [28] and Annexin A1 is among other proteins in exosomes derived from apical, but not basolateral, plasma membrane of mouse collecting duct cells [29].
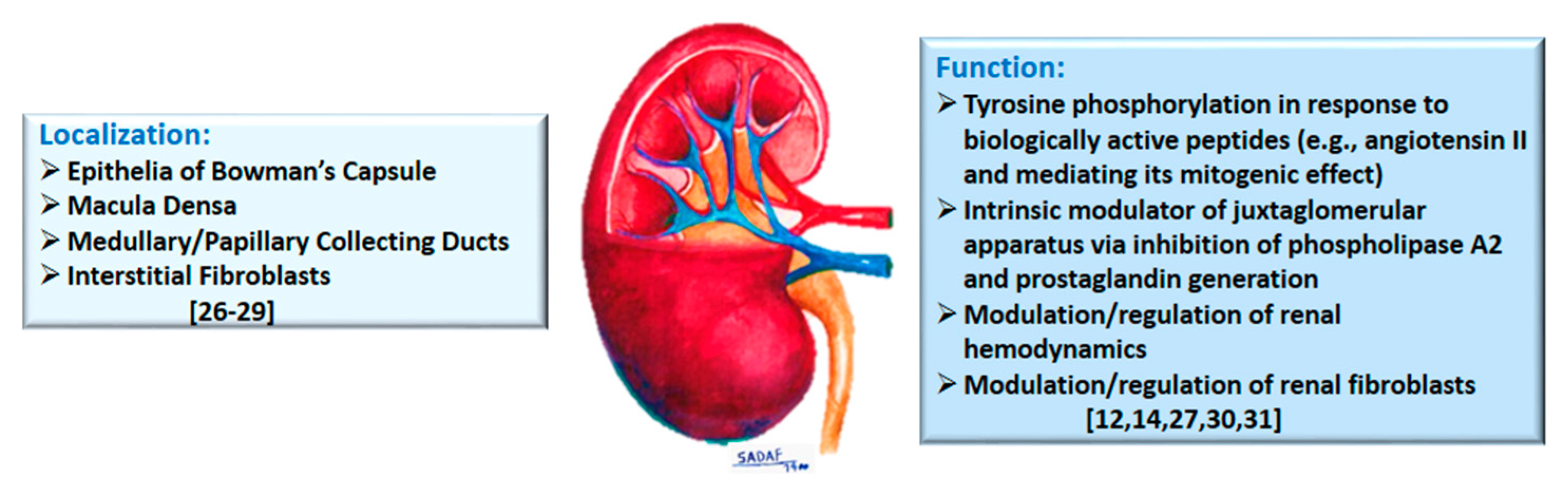
Figure 1. The diagram summarizes localization and physiological functions attributed to Annexin A1 in the kidney; relevant citations are provided in brackets.
The discovery of annexins in the kidney, and their differential localization, led to studies focused on determination of its functional roles. Accordingly, mesangial cell cultures, constitutively expressing annexin 1, alone, were shown to incorporate 32P with (tyrosine) phosphorylation being upregulated (2–3 fold) upon exposure to epidermal growth factor, angiotensin II, arginine-vasopressin or endothelin I [27]. It is suggested that annexin 1 tyrosine phosphorylation, by the aforementioned biologically active peptides, occurs via G-protein-linked receptors and that a plausible role exists for annexin 1 in the mitogenic effect of angiotensin II, arginine-vasopressin, and endothelin I in rat glomerular mesangial cells. A subsequent study investigated the role of annexin 1 in the response of mesangial cells to DEXA and/or interleukin (IL)-1β in relation to (14 kDa Group II) PLA2 activity (Figure 1). Mesangial cells annexin 1 remained unchanged despite marked increases in PLA2 activity and protein levels in response to IL-1β and DEXA. In addition, DEXA did not induce translocation of annexin 1 from the cytosol to cell membrane or its secretion. It was concluded that DEXA-induced inhibition of PLA2 in rat mesangial cells is due to the suppression of its gene expression rather than being mediated via annexin 1 [30]. However, utilizing annexin 1-deficient U937 cells (e.g., used in monocyte differentiation studies) and mice lacking ANXA1 gene, a clear role for Annexin A1 in the inhibition of cytosolic PLA2 has been suggested [12][14].
More recently, the recognition of expression of Annexin A1 in cells of the juxtaglomerular apparatus prompted the determination of its role in relation to cyclooxygenase-2 (COX-2) in rat [31]. Juxtaglomerular apparatus is formed by the glomerular afferent arteriole and the distal convoluted tubule and regulates glomerular filtration rate, solute transport, and blood pressure. Results indicated that (a) Annexin A1 is extensively co-localized with COX-2 in macula densa, (b) furosemide (a high ceiling diuretic and COX2 inducer) increased the expression of both COX-2 and Annexin A1 (which were highly co-localized), thereby suggesting that Annexin A1 expression is regulated by Na+/K+/2Cl− cotransporter, (c) Annexin A1 deficient mice displayed greater COX-2 positive cells in macula densa cells along with greater juxtaglomerular renin secretion (normalized to glomerular number), (d) cultured macula densa cells, transfected with full-length rat Annexin A1, displayed downregulation of mRNA for COX-2, (e) DEXA treatment induced Annexin A1 mRNA expression and protein secretion and suppressed COX-2 mRNA, and (f) cyclosporine H (0.1μM)-induced inhibition of Fpr1 attenuated the effect of DEXA on COX-2 suppression in the macula densa while the Fpr2/Fpr3 antagonist, WRW4, had no effect. Based on the collective observations, it is concluded that Annexin A1 inhibits COX-2 in macula densa, with consequent inhibition of PGE2 synthesis, thereby serving as an intrinsic modulator of juxtaglomerular apparatus [31]. Thus, aside from inhibition of PLA2, it appears that Annexin A1 also inhibits COX2 with subsequent downregulation of prostanoids.
In conclusion, given the pivotal role of the juxtaglomerular apparatus in the regulation of glomerular filtration, sodium reabsorption, and blood pressure control, unraveling of the physiological role of Annexin A1 in the kidney is of major relevance and significance for homeostasis. In addition, mesangial cells expression of Annexin A1 is of importance given the role of these cells in maintaining structural integrity of glomerular microvascular bed and mesangial matrix homeostasis as well as a myriad of responses to hemodynamic and immunologic injuries (e.g., proliferation, hypertrophy, matrix synthesis and degradation, production of cytokines and growth factor, oxidative stress and apoptosis) [32], aspects that require further investigation.
3. Role of Annexin A1 in Pathologies of the Kidney
3.1. Annexin A1 and Immune/Inflammatory Responses in Kidney Pathologies
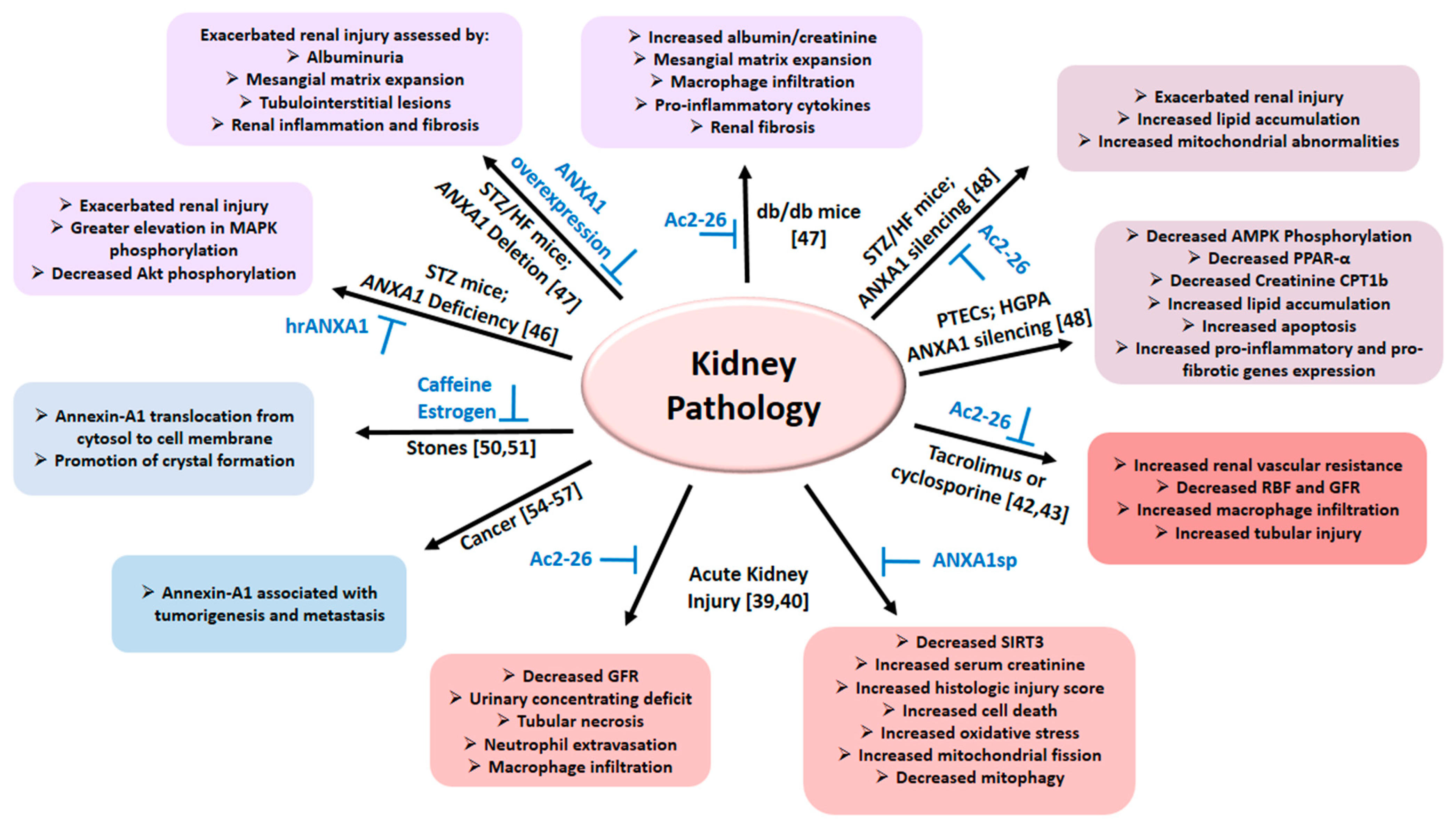
Figure 2. The diagram summarizes the role of Annexin A1 in various pathologies of the kidney as detailed in the text; relevant citations are provided in brackets. Abbreviations: AMPK: Adenosine 5′-monophosphate-activated protein kinase; ANXA1: Annexin A1; Ac2-26: Annexin A1 peptide mimetic; ANXA1sp: Annexin A1 tripeptide mimetic; CTP1b: Carnitine palmitoyltransferase 1b; GFR: Glomerular filtration rate; HFD: High fat diet; HGPA: High glucose palmitic acid; PPAR-α: Proliferator-activated receptor-α; PTECs: Proximal tubule epithelial cells; RBF: Renal blood flow; Sirtuin-3 (SIRT3); STZ: Streptozotocin.
Table 1. Annexin A1 as a diagnostic/prognostic biomarker.
| Condition | Finding | Citation(s) |
|---|---|---|
| Lupus Nephritis | Increased serum IgG2 against Annexin A1 | [33] |
| Glomerulonephritis with polyangiitis | Increased Annexin A1 expression in neutrophils | [34] |
| Kidney injuries of diverse pathogenesis (e.g., diabetes mellitus, pyemic secondary AKI, Adriamycin-induced nephrotoxicity, etc.) | Increased kidney, serum (or plasma) and/or urinary Annexin A1 | [35][36][37][38][39][40] |
| Patients with acute heart failure associated with impaired kidney function | Increased plasma Annexin A1 associated with worse congestion, morbidity and mortality Annexin A1 is a Potential Novel Biomarker of Congestion in Acute Heart Failure. | [41] |
| Renal cancer | Marker of poor prognosis Predictor of poor sunitinib response | [42][43][44][45] |
The early recognition that Annexin A1 reduces leucocyte migration, in response to cytokines and chemokines in animal models of inflammation, led to the comparison of its temporal effects to those of DEXA in the lipopolysaccharide (LPS)-induced inflammatory response in rat [46]. Accordingly, LPS superfusion resulted in the adhesion of leukocytes to the endothelium followed by increased emigration from post-capillary venules (over 2 h), effects which were attenuated by either Annexin A1 or DEXA. The activity of myeloperoxidase (MPO) was increased in the kidney, lung, and ileum after 6-h of exposure to LPS, thereby suggestive of neutrophil accumulation in these tissues. DEXA reduced LPS-induced elevation in MPO activity in the ileum but not in the lungs or kidneys, thereby suggesting differential responses of vascular beds in the ileum than those of the lung or the kidney. However, Annexin A1 did not affect LPS-induced increase in MPO activity in any of the aforementioned tissues. It is concluded that Annexin A1 reduces leukocyte adhesion and emigration following a short-term (2 h), but not a longer-term (6 h) exposure to LPS. Therefore, it is noteworthy that a mechanistic link between Annexin A1 and hydrogen sulfide pathway has been reported in leukocyte trafficking in experimental models of inflammation (e.g., induced by LPS) [47].
The potential role of Annexin A1 in relation to pathogenesis of the autoimmune disease of lupus nephritis (LN) has been investigated using renal biopsy and blood samples of patients afflicted with the disease [33]. Accordingly, microdissected glomeruli from biopsy samples of patients with LN were subjected to proteomic analyses. In addition, serum samples from large cohorts of patients with systemic lupus erythematosus, with and without LN, and glomerulonephritis of other etiologies were tested. Glomerular IgGs recognized 11 podocyte antigens although reactivity varied depending on LN pathology. Interestingly, IgG2 autoantibodies against Annexin A1, and α-enolase, were predominant and identified in 10 and 11 of the 20 biopsy samples, respectively. α-enolase is a metabolic enzyme but also serves multiple other functions (e.g., cell surface receptor for plasminogen). Immunohistochemical analyses revealed co-localization of Annexin A1 or α-enolase with IgG2 in glomeruli. In addition, high levels of serum anti-α-enolase IgG2 and/or anti-Annexin A1 IgG2 were detected in most patients with LN but not patients with other forms of glomerulonephritis. Serum levels of both autoantibodies decreased significantly after a year of LN therapy. Anti-α-enolase IgG2 recognized specific epitopes of α-enolase and did not cross-react with dsDNA. Although circulating dsDNA antibodies (which can interact with antigens on cell membranes of podocytes and mesangial cells) have been considered to play an important pathogenic role in LN, more recent information indicates that anti-DNA deposits account for 10–20% of IgG, thereby adding further relevance to findings. Furthermore, monoclonal IgG2 derived from lupus-prone MRL-lpr/lpr mice recognized human α-enolase, suggesting homology between animal models and human LN. It was concluded that LN manifests a multi-antibody composition and that IgG2 autoantibodies against α-enolase and Annexin A1 predominate in the glomerulus and can also be detected in serum. Thus, given the increased predictive power of a combination of markers, the development of a panel composed of multiple antibodies (e.g., targeting Annexin A1, α-enolase, etc.) could serve as a useful surrogate biomarker of LN.
More recently, the role of Annexin A1, in the context of its cell death/survival function, has been reported in nephropathy associated with the autoimmune disease of glomerulonephritis with polyangiitis (GPA) [34]. This condition is associated with antinuclear-cytoplasmic antibodies (ANCA) targeting proteinase-3, thereby causing kidney injury. Indeed, increased neutrophil membrane proteinase-3 is a feature of apoptosis in this condition. Accordingly, proteomic analyses were carried out for comparison of neutrophils of patients with GPA and those of healthy controls under baseline condition and during apoptosis. It was reported that neutrophils proteome was markedly different at the onset of GPA and that this alteration became more pronounced following ex vivo apoptosis. Alterations in cell death/survival proteins were noted in neutrophils of patients with GPA including proteinase-3 binding proteins such as Annexin A1 (as well as calreticulin, and phospholipid scramblase 1). Indeed, increased membrane expression of Annexin A1 was a feature of apoptosis. It was proposed that the expression of these proteins at the cell membrane of apoptotic neutrophils, of patients with GPA but not healthy subjects, account for persistent inflammation and autoimmunity. Thus, differential expression of these proteins offers the opportunity for novel biomarker discovery and therapeutic option(s) for the management of kidney injury in GPA.
In conclusion, the immune system contributes to kidney damage that can be of long-term sequela and progression to chronic kidney disease. Autoantibodies against renal antigens (e.g., collagen IV in glomerular basement membrane) account for direct immune-mediated renal injury while immune complex formation, in systemic forms of autoimmunity, contribute to indirect immune-mediated kidney injury. The ensuing dysregulated inflammatory responses cause exacerbated recruitment of immune cells to the site of injury, which contribute to the pathogenesis of chronic kidney disease and, if left uninterrupted, to end-stage renal failure [48]. Given the multifaceted role of Annexin A1 in various inflammatory pathologies, the availability of its peptide and small molecule modulators [49] paves the way for establishing pathogenic mechanisms and potential therapeutic options for these conditions.
3.2. Annexin A1 and Acute Kidney Injury
An early study reported marked increase in Annexin A1 level in homogenates of the whole kidney two days after an ischemic insult. Further, immunohistochemical analyses showed restricted expression in some segments similar to non-ischemic kidneys (as alluded to earlier) but the thick ascending limb stained heavily following an ischemic insult suggestive of altered expression of Annexin A1 as a pathophysiological response [26]. Thereafter, the impact of an Annexin A1 mimetic peptide (Ac2-26) was explored in rat kidneys subjected to ischemia-reperfusion injury- a model of acute kidney injury (AKI) [50]. Administration of Ac2-26 before clamping of the renal artery conferred marked renoprotection as indexed by restoration of glomerular filtration rate and urine osmolality and prevented acute tubular necrosis. Further, the treatment reduced neutrophil extravasation and macrophage infiltration but did not influence lymphocyte movement. Interestingly, the ischemia-reperfusion insult increased Annexin A1 expression in renal epithelial cells, an effect that was attenuated by Ac2-26 administration. In addition, isolated proximal tubules suspension displayed reduced hypoxic/reoxygenation injury in response to Ac2-26. It is concluded that while neutrophils are important mediators of renal injury in response to an ischemia-reperfusion insult, Annexin A1 is an important regulator of the defense response of epithelial cells in this condition and it may serve as a novel therapeutic option for relevant clinical conditions such as renal transplantation [50].
More recently, a rather detailed study examined the impact of an Annexin A1 tripeptide mimetic (ANXA1sp) on the outcome of AKI in the context of assessment of sirtuin-3 (SIRT-3) and mitochondrial function [51]. The focus on SIRT-3 relates to its ability to regulate pivotal cellular processes, including mitochondrial function and bioenergetics, mitophagy, and oxidative stress. Accordingly, utilizing the murine model of AKI, induced by renal ischemia reperfusion injury, ANXA1sp was administered an hour before renal vessel clamping and an hour after restoration of renal blood flow. Treatment with ANXA1sp reduced kidney damage as indexed by histologic injury score and serum creatinine level. In addition, it reduced cell death (TUNEL staining) in association with inhibition of oxidative stress and promotion of mitochondrial integrity and function (indexed by assessment of superoxide dismutase, 8-hydroxydeoxyguanine, oxyguanosine DNA glycosylase, mitochondrial transcription factor A) in the ischemic-reperfused kidneys. Similarly, ANXA1sp reduced cell death in HK-2 cells subjected to hypoxia. The impact of ANXA1sp on other markers of mitochondrial integrity and function were as follows. While ANXA1sp reduced dynamin-related protein-1 level (marker of mitochondrial fission) in ischemic-reperfused kidneys, suggestive of curtailing mitochondrial fragmentation, the treatment improved markers of mitophagy (e.g., LC3II, PTEN induced putative kinase 1 (PINK1), and Parkin) under these conditions. Importantly, ANXA1sp increased mRNA and protein expressions of SIRT3 in ischemic-reperfused kidneys compared to their vehicle-treated counterparts (but these parameters remained lower than those of their sham-operated controls). Further, in vitro studies showed that SIRT3 silencing reverses protection against hypoxic cell death conferred by ANXA1sp. It is concluded that renoprotective effects of ANXA1sp relate to upregulation of SIRT3 and that it offers therapeutic potential for limiting kidney injury as a perioperative agent in conditions associated with renal ischemia (e.g., surgery and transplantation) [51].
In another study, serum Annexin A1 was used as an index of success of continuous renal replacement therapy (CRRT) in the setting of pyemic secondary acute kidney injury (AKI). Annexin A1 increased in this condition and its peak level in the CRRT-responder group decreased more rapidly compared to the non-responder group. Interestingly, however, other indices of kidney function (e.g., creatinine, urea and nitrogen levels) did not distinguish responders vs. non-responders to CRRT. It is concluded that serum Annexin A1 level predicts the outcome of CRRT on pyemic secondary AKI [35].
In conclusion, AKI remains a major clinical problem as it affects critically ill patients and has detrimental effects on both short- and long-term outcomes. AKI, secondary to ischemia-reperfusion injury, is associated with several clinical conditions, including renal transplantation, partial nephrectomy, cardiac surgery, shock, and repair of some abdominal aneurysms. Multiple and diverse pathogenic mechanisms contribute to AKI including dysregulation of inflammatory mechanisms as both triggering and essential amplifying factors leading to an excessive inflammatory response thereby exacerbating tissue injury. Given the role of Annexin A1 in regulating multiple components of the immune and inflammatory responses, further investigation of its potential beneficial effects in such diverse conditions is warranted. Moreover, investigation of the impact of Annexin A1, or its mimetics, i.e., peptides and small molecules, initiated at various time points after the restoration of renal blood flow and determination of short- and long-term outcomes represent fertile ground for future investigation.
3.3. Annexin A1 and Immunosuppressive Agents
Despite the revolutionary impact of immunosuppressive agents on organ transplantation, nephrotoxicity (e.g., impaired renal hemodynamics) is a major adverse outcome associated with use of agents such as cyclosporine or tacrolimus. The administration of tacrolimus to rats impaired renal function manifested as increased renal vascular resistance and reductions in renal blood flow and glomerular filtration rate associated with increased macrophage infiltration and tubulointerstitial injury; these effects were accompanied with increased renal tissue expression of Annexin A1. Further, the administration of Ac2-26 attenuated tacrolimus-induced dilatation of renal tubules and macrophage infiltration, effects that could be of therapeutic value [36]. Thereafter, this group explored the impact of Ac2-26 in cyclosporine-treated rats [37]. Accordingly, they showed that cyclosporine treatment causes significant impairment of renal blood flow and glomerular filtration rate along with renal tubule dilatation and macrophage infiltration accompanied with increased kidney expression of Annexin A1. Treatment with Ac2-26 ameliorated renal hemodynamic changes, tubular damage, and macrophage infiltration in cyclosporine-treated animals. Despite these promising observations, their relevance to kidney dysfunction/injury in human subjects receiving cyclosporine or tacrolimus therapy (e.g., organ transplantation or autoimmune disorders) remains to be established.
3.4. Annexin A1 and Renal Injury of Diverse Etiopathogenesis
The observation of upregulation of Annexin A1 in kidney injury models (e.g., AKI and cyclosporine- and adriamycin-induced nephrotoxicity) has led to the determination of whether urinary level of Annexin A1 can serve as an index of glomerular injury in adriamycin-induced nephrotoxicity or for a heterogeneous groups of patients manifesting minimal change disease (MCD) vs. those with other forms of kidney pathologies (i.e., IgA nephropathy, membranous glomerulonephritis, focal segmental sclerosis, diabetic nephropathy, lupus nephritis, and crescentric glomerulonephritis) [38]. MCD refers to glomerular injury detected via electron microscopy and is the etiology of nephrotic syndrome in 10–25% of human subjects. It was reported that (a) urinary excretion of Annexin A1 is detectable in both the animal model of adriamycin-induced nephrotoxicity and in patients except those with MCD, (b) urinary Annexin A1 was positively correlated with various types of glomerular lesions, and (c) urinary Annexin A1 was detected in diabetic patients with otherwise normal albumin excretion. It is concluded that Annexin A1 may serve as an early diagnostic and prognostic marker of glomerular injury and may differentiate patients with MCD vs. those with other pathologies of glomeruli [38].
3.5. Annexin A1 and Renal Fibrosis
Given the anti-inflammatory effects of Annexin A1, and its interaction with Fpr2, in protection against organ fibrosis, the role of Annexin A1 in kidney fibroblasts has been examined utilizing an animal model of renal fibrosis induced by inhibiting angiotensin II type 1 receptor with L-158.809 during the nephrogenic period [28]. The treatment apparently causes late-onset hypertensive nephropathy and fibrosis. In addition, effects of Annexin A1 were examined in cultured renal fibroblasts obtained from Annexin A1 knockout and wild type mice. The renal fibrotic model displayed matrix foci containing CD73+ fibroblasts, alpha-smooth muscle actin (α-SMA+) myofibroblasts and CD68+ macrophages. Further, in these animals, transforming growth factor-β (TGF-β) and Annexin A1 mRNAs were increased compared to those of controls. Annexin A1 expression was identified in macrophages and fibroblasts (with intense staining); however, myofibroblasts were negative. On the other hand, Fpr2 was expressed by fibroblasts, myofibroblasts, macrophages and endothelial cells. Further, Annexin A1 and Fpr2 immunoreactivity were increased in fibrotic foci, with fibroblasts and macrophages expressing both proteins. Renal fibroblasts obtained from Annexin A1 knockout animals displayed markedly higher α-SMA and collagen 1A1 mRNA levels than controls, with the effect much more pronounced for collagen 1A1 mRNA. Treatment of renal fibroblasts, obtained from wild type mice, with TGF-β increased mRNA levels of α-SMA and collagen 1A1. These changes were markedly attenuated in response to Annexin A1 overexpression. It is concluded that ubiquitous expression of Annexin A1 and Fpr2 in the kidney interstitium regulates functional phenotype of renal fibroblasts and their contribution to synthesis of extracellular matrix proteins. Thus, it is likely that activation of annexin A1/Fpr2 signaling in fibroblasts could exert antifibrotic impact in chronic kidney disease. In this context, it is noteworthy that Annexin A1 gene expression is upregulated in renal cortex of the deoxycorticosterone-acetate/NaCl rat model of hypertension [39], a condition associated with marked kidney inflammation, fibrosis and dysfunction; increased Annexin A1 expression likely serves as a compensatory mechanism curtailing fibrosis.
3.6. Annexin A1 and Metabolic Diseases
Microvascular complications involving major target organs (e.g., kidney and heart) are common in diabetes mellitus for which inflammatory mechanisms play major pathogenic roles. Thus, the role of Annexin A1, as an endogenous anti-inflammatory mediator, has been examined in cardiorenal complications of type 1 diabetic patients and the streptozotocin (STZ)-induced diabetic mouse model [40]. Patients with type 1 diabetes, irrespective of nephropathy, displayed increased plasma Annexin A1. However, Annexin A1 level was reduced in renal and cardiac tissue lysates of diabetic animals. Further, Annexin A1 deficiency was associated with more severe STZ-induced diabetes accompanied with more severe nephropathy (indexed by albumin-to-creatinine ratio, glomerular area and fibrosis) and cardiomyopathy (indexed by ejection fraction, fractional shortening, etc.). To provide mechanistic linkage, it was shown that Annexin A1 knockdown in non-diabetic mice significantly increases phosphorylation of several MAPK (i.e., p38, ERK and JNK) in the heart and kidney, an effect that further increased with co-existence of STZ-induced type 1 diabetes. While prophylactic treatment of type 1 diabetic mice with human recombinant Annexin A1 (hrANXA1) reduced kidney and heart abnormalities, treatment after development of significant cardiorenal abnormalities in diabetic mice prevented further decline in cardiac and renal functions. In addition, treatment with hrANXA1 reduced the elevation in phosphorylation of MAPK and restored phosphorylation status of Akt in diabetic mice. It was concluded that increased plasma Annexin A1 occurs in type 1 diabetic subjects independent of significant impairment of kidney function. In addition, hrANXA1 protected against cardiorenal abnormalities of the murine model of type 1 diabetes via inhibition of MAPK but restoration of pro-survival activity of Akt [40]. Thus, Annexin A1 may represent a novel therapeutic option for microvascular complications of diabetes mellitus.
With respect to diabetes mellitus, increased renal Annexin A1 is shown to correlate with kidney function and outcomes in established diabetic nephropathy in a human cohort [52]. According, using the STZ-injected/high fat-fed (STZ/HF) mice, it was shown that Annexin A1 deficiency exacerbated renal injury manifested as exacerbated albuminuria, mesangial matrix expansion, tubulointerstitial lesions, kidney inflammation and fibrosis. On the other hand, Annexin A1 overexpression attenuated renal injury in diabetic mice. Further, treatment with Ac2-26 attenuated kidney injury in db/db mice (a genetic model of type 2 diabetes) and in diabetic Annexin A1 knockout mice. Mechanistic studies revealed that intracellular Annexin A1 binds to the p65 subunit of nuclear factor-κB (NF-κB), thereby inhibiting its activity and reducing inflammation [52]. Thus, Annexin A1 may serve as a therapeutic option to ameliorate diabetic nephropathy.
The role of Annexin A1 in diabetic nephropathy has been further investigated in the context of assessment of associated metabolic abnormalities [53]. Deletion of Annexin A1, in STZ/HF mice, exacerbated renal injury indexed by worsening of albuminuria, mesangial matrix expansion and tubulointerstitial injury. Further, Annexin A1 deficiency increased intra-renal accumulation of lipids and mitochondrial abnormalities. Treatment with Ac2-26 exerted beneficial effects against lipid accumulation in diabetic mice. Silencing of ANXA1 in high-glucose/palmitic acid (HGPA)-treated proximal tubule epithelial cells (PTECs) suppressed phosphorylated Thr172 adenosine 5′-monophosphate-activated protein kinase, culminating in decreased expression of proliferator-activated receptor-α and carnitine palmitoyltransferase 1b accompanied with increased HGPA-induced lipid accumulation, apoptosis and increased expression of pro-inflammatory and pro-fibrotic genes. Further, renal biopsies of diabetic nephropathy patients displayed more intense staining for adipose differentiation-related protein (ADRP) than healthy controls with a negative correlation between ADRP and estimated glomerular filtration rate. Interestingly, tubulointerstitial Annexin A1 expression correlated with ADRP suggestive of its association with lipid abnormalities of diabetic nephropathy patients. It was concluded that Annexin A1 regulates lipid metabolism (e.g., of PTECs) to ameliorate disease progression, and that its modulation likely offering therapeutic potential for diabetic nephropathy.
In conclusion, dysregulation of Annexin A1 is involved in renal conditions of diverse etiopathogenesis. Interestingly, and as alluded to above, increased kidney expression of Annexin A1 is a feature of some of these conditions likely suggestive of serving as compensatory mechanism to curtail tissue injury (Table 1). Importantly, however, Annexin A1 can be cleaved, at the N-terminal domain, by a number of enzymes including elastase, proteinase 3, metalloproteinases, cysteine protease, calpain I or aspartyl protease cathepsin D among others [7]. Interestingly, products of some of these biotransformation pathways (e.g., by calpain or cathepsin D) are believed to exert pro-inflammatory effects thereby interfering with Annexin A1′s anti-inflammatory and pro-resolving activity [7]. This likely explains why compensatory increase in Annexin A1 in some of the aforementioned conditions may be insufficient to curtail inflammation while treatment with Annexin A1 mimetics restores tissue homeostasis. Nonetheless, while Annexin A1 mimetic peptides (e.g., Ac2-26) serve as valuable experimental tools, the development of orally bioavailable agonists of Annexin A1 receptors (i.e., Fpr1/2) would help better establish therapeutic values of modulation of Annexin A1 signaling.
3.7. Annexin A1 and Kidney Stone
Elevation in urinary calcium level is associated with impaired kidney function and kidney stone formation. Thus, proteomic analysis of renal tubular cells was carried to determine whether their protein profile is altered upon exposure to a high calcium medium [54]. Accordingly, it was shown that Madin Darby Canine Kidney (MDCK) cells exposed to high calcium displayed increased capacity for binding to calcium oxalate monohydrate crystals accompanied with increased Annexin A1 expression, as a calcium-binding protein, on the apical surface of the cells, an effect reversed by pre-treatment with antibody targeting Annexin A1. Importantly, exposure to high calcium also led to impaired wound healing and cell proliferation. It was concluded that these in vitro observations implicate a role for Annexin A1 in nephrolithiasis and kidney dysfunction that may be of relevance to hypercalciuria and kidney stone formation.
A subsequent study further explored the role of Annexin A1 in kidney stone formation in the context of an assessment of the effects of caffeine in this process given reports that caffeinated beverages reduce the risk of kidney stone formation [55]. Caffeine reduced calcium oxalate monohydrate crystal numbers but increased crystal size thereby resulting in no change in the size of the crystal mass. Interestingly, however, caffeine treatment, dose-dependently, reduced the ability of MDCK cells to bind crystals, an effect associated with decreased expression of Annexin A1 on the apical surface suggestive of its translocation into the cytoplasm; no change was reported for several other calcium oxalate monohydrate crystal binding proteins (i.e., annexin-A2, α-enolase, HSP70, and HSP90). Further, caffeine reduced intracellular Ca2+ level but increased the calcium secretory index. It is concluded that caffeine exerts a protective effect against kidney stone formation likely via the translocation of Annexin A1 from the apical membrane of cells into the cytosolic compartment, thereby reducing the crystal-binding capacity of renal tubular epithelial cells. In a more recent study, these investigators explored the role of Annexin A1 in the purported protective effect of estrogen on kidney stone formation given reports of lower risk in female than male [56]. Utilizing MDCK cells coupled with proteomic analyses, it is reported that seven-day exposure to estrogen resulted in the differential expression of 58 proteins which are primarily involved in functional networks related to “binding and receptor,” “metabolic process,” and “migration and healing”. Further, estrogen-treated cells displayed reduced calcium oxalate crystal-binding capacity coupled with decreased cell membrane levels of Annexin A1 and α-enolase (as calcium oxalate crystal-binding receptors). Interestingly, estrogen reversed the elevations in cell surface expression of Annexin A1, and α-enolase, secondary to high-calcium and high-oxalate challenge. Estrogen also decreased intracellular ATP level and enhanced cell migration and tissue healing. Thus, it is likely that estrogen promotes cellular processes that favor reduced cell surface expression of receptors for calcium oxalate crystals and enhanced cell proliferation and tissue healing, thereby contributing to prevention of stone formation.
In conclusion, nephrolithiasis is often a painful condition with its prevalence increasing worldwide. The annual incidence is estimated to be about eight per 1000 adults that peaks about midlife in developed countries. Most cases of kidney stones are of noninfectious etiologies but are associated with environmental factors (climate and fluid intake) and several comorbidities including metabolic syndrome/obesity, excessive protein intake and gout. Shock wave therapy and surgery are among management approaches but patients experience recurrent stones within 10 years after removal [55]. Thus, aside from preventive measures (e.g., hydration), attention is increasingly focused on modulation of crystal formation, growth and aggregation to decrease or prevent their adhesion to renal epithelial cells. The studies focused on the role of Annexin A1 in pathogenesis of nephrolithiasis suggest that downregulation of renal cell membrane Annexin A1 may offer a promising approach. Thus, the development of therapeutically useful inhibitors of Annexin A1 is a step in that direction.
3.8. Annexin A1 and Renal Cancer
The role of Annexin A1 has been extensively studied in a number of cancers and tumors cell lines [57]. With respect to kidney cancer, early recognition of Annexin A1-mediated growth inhibition of glucocorticoids in human lung adenocarcinoma cell line led to the determination of the impact of hydrocortisone and hrANXA1 on cultured human mesangial cells (CHMC) [58]. Annexin A1 dose-dependently inhibited proliferation of CHMC (unstimulated or activated with platelet-derived growth factor) as assessed by 3H-thymidine incorporation and cell count. Similarly, hydrocortisone dose-dependently inhibited CHMC proliferation. Treatment with an antibody against Annexin A1 reversed hydrocortisone-induced inhibition of CHMC proliferation. Further studies using flow cytometry and indirect immunofluorescent microscopy indicated specific Annexin A1 binding sites on the surface of CHMC. It was concluded that corticosteroids cause generation/secretion of Annexin A1 by CHMC that, in turn, results in suppression of their proliferation via specific binding sites on CHMC.
The role of Annexin A1, in conventional renal cell carcinoma (CRCC), without or with metastasis, was investigated with respect to tumorigenesis, metastatic potential and clinical outcome [42]. Immunohistochemical staining was correlated with Fuhrman nuclear grade (an independent predictor of cancer-specific survival), amount of eosinophilic cells and higher clinical stage leading to the conclusion that that Annexin A1 could serve as a prognostic marker for tumor progression. Another study assessed cytoplasmic staining of Annexin A1 in metastatic renal cell carcinoma (mRCC; 77 cases) treated with nephrectomy followed by therapy with sunitinib (a small molecule inhibitor of receptor tyrosine kinase) [43]. Annexin A1 negative cases (32%) showed better progression-free survival and overall survival than those cases with positive cytoplasmic Annexin A1 staining. It was concluded that cytoplasmic expression of Annexin A1 was a negative predictive marker for sunitinib treatment in mRCC patients, likely because its anti-angiogenic effect cannot overcome the pro-angiogenic effect of Annexin A1.
More recently, assessment of Annexin A1 expression in renal cell carcinoma of 27 patients was coupled with the use of log-rank test for assessment of disease-free survival [44]. Accordingly, 48% of cases displayed high Annexin A1 expression in cell membranes of tumor cells while others showed weak or negligible staining. Further, patients whose tumors stained intensely for Annexin A1 had significantly poorer disease-free survival than others. Knockdown of Annexin A1 in Caki-2 cells (a human clear cell renal cell carcinoma line) resulted in reduced proliferation, invasion, motility and adhesion compared to control cells. Further, Annexin A1 deficient cells expressed lower relative levels of membrane-type 1 matrix metalloproteinase and hypoxia-inducible factor 1-α transcripts. It was concluded that Annexin A1 is associated with the malignant potential of renal cell carcinoma and, thus, could serve as a marker of poor prognosis.
In addition to context-specific tumorigenesis, many other functions have been attributed to Annexin A1 including clearance of apoptotic cells [45]. Accordingly, expression of Annexin A1 was assessed in fetal kidneys (at different gestational periods), mature kidneys and in kidney cancer tissues to gain insight into its localization and its potential role during nephrogenesis and in renal tumors. Annexin A1 was expressed in mesangial cells and podocytes of developing glomeruli and renal cortex of fetal kidneys (days 14–19 of gestation); mesangial cells Annexin A1 expression declined over time but persisted into adulthood. Importantly, Annexin A1 expression increased with the progression of clear cell renal cell carcinoma (and other cancer types), suggestive of its potential role in tumorigenesis. It was concluded that Annexin A1 plays important functional roles in podocytes and mesangial cells, as integral components of renal glomeruli, and that their unraveling could help with the development of diagnostic, prognostic, and therapeutic strategies for various renal pathologies.
In conclusion, Annexin A1 has been implicated in cell proliferation, apoptosis, sensitivity to chemotherapy as well as invasion and metastasis, aspects that are essential in cancer biology. Nonetheless, it is suggested that Annexin A1 may serve as a partial functional mediator of tumorigenesis and metastasis rather than a tissue-specific mediator for predicting cancer initiation and/or metastasis [57]. With respect to renal cancer, and as described above, while the inhibitory effect of Annexin A1 on the proliferation of mesangial cells has been reported, other studies suggest tumorigenesis and malignant potentials. Thus, the development and use of effective inhibitors of Annexin A1 signaling would help better understand the role of Annexin A1 in tumor biology.
4. Potential Use of Annexin A1 as a Biomarker
Several aforementioned studies have indicated the potential use of Annexin A1 as a diagnostic and/or prognostic biomarker (Table 1). In addition, a recent study concluded that elevated plasma Annexin A1 level is associated with worse congestion, higher risk for further elevation of creatinine, and higher rates of 60-day morbidity or all-cause mortality and 180-day all-cause mortality in hospitalized patients with acute heart failure associated with impaired kidney function (Table 1) [41].
This entry is adapted from the peer-reviewed paper 10.3390/cells10123420
References
- Moss, S.E.; Morgan, R.O. The annexins. Genome Biol. 2004, 5, 219.
- Cantacessi, C.; Seddon, J.M.; Miller, T.L.; Leow, C.Y.; Thomas, L.; Mason, L.; Willis, C.; Walker, G.; Loukas, A.; Gasser, R.B.; et al. A genome-wide analysis of annexins from parasitic organisms and their vectors. Sci. Rep. 2013, 3, 2893.
- Gerke, V.; Moss, S.E. Annexins: From Structure to Function. Physiol. Rev. 2002, 82, 331–371.
- Markoff, A.; Gerke, V. Expression and functions of annexins in the kidney. Am. J. Physiol.-Renal Physiol. 2005, 289, F949–F956.
- Cloix, J.F.; Colard, O.; Rothhut, B.; Russo-Marie, F. Characterization and partial purification of ‘renocortins’: Two polypeptides formed in renal cells causing the anti-phospholipase-like action of glucocorticoids. Br. J. Pharmacol. 1983, 79, 313–321.
- Morgan, R.O.; Fernandez, M.P. Annexin gene structures and molecular evolutionary genetics. Cell. Mol. Life Sci. 1997, 53, 508–515.
- Araújo, T.G.; Mota, S.T.S.; Ferreira, H.S.V.; Ribeiro, M.A.; Goulart, L.R.; Vecchi, L. Annexin A1 as a Regulator of Immune Response in Cancer. Cells 2021, 10, 2245.
- Sheikh, M.H.; Solito, E. Annexin A1: Uncovering the Many Talents of an Old Protein. Int. J. Mol. Sci. 2018, 19, 1045.
- Solito, E.; Mulla, A.; Morris, J.F.; Christian, H.C.; Flower, R.J.; Buckingham, J.C. Dexamethasone Induces Rapid Serine-Phosphorylation and Membrane Translocation of Annexin 1 in a Human Folliculostellate Cell Line via a Novel Nongenomic Mechanism Involving the Glucocorticoid Receptor, Protein Kinase C, Phosphatidylinositol 3-Kinase, and Mitogen-Activated Protein Kinase. Endocrinology 2003, 144, 1164–1174.
- Perretti, M.; D’Acquisto, F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 2009, 9, 62–70.
- Croxtall, J.D.; Choudhury, Q.; Newman, S.; Flower, R.J. Lipocortin 1 and the control of cPLA2 activity in A549 cells: Glucocorticoids block EGF stimulation of cPLA2 phosphorylation. Biochem. Pharmacol. 1996, 52, 351–356.
- Solito, E.; Raguenes-Nicol, C.; De Coupade, C.; Bisagni-Faure, A.; Russo-Marie, F. U937 cells deprived of endogenous annexin 1 demonstrate an increased PLA2activity. Br. J. Pharmacol. 1998, 124, 1675–1683.
- Parente, L.; Solito, E. Annexin 1: More than an anti-phospholipase protein. Inflamm. Res. 2004, 53, 125–132.
- Hannon, R.; Croxtall, J.D.; Getting, S.J.; Roviezzo, F.; Yona, S.; Paul-Clark, M.J.; Gavins, F.N.E.; Perretti, M.; Morris, J.F.; Buckingham, J.C.; et al. Aberrant inflammation and resistance to glucocorticoids in Annexin 1−/− Mouse. FASEB J. 2002, 17, 253–255.
- Vago, J.P.; Tavares, L.P.; Riccardi, C.; Teixeira, M.M.; Sousa, L.P. Exploiting the pro-resolving actions of glucocorticoid-induced proteins Annexin A1 and GILZ in infectious diseases. Biomed. Pharmacother. 2021, 133, 111033.
- Ricci, E.; Ronchetti, S.; Pericolini, E.; Gabrielli, E.; Cari, L.; Gentili, M.; Roselletti, E.; Migliorati, G.; Vecchiarelli, A.; Riccardi, C. Role of the glucocorticoid-induced leucine zipper gene in dexamethasone-induced inhibition of mouse neutrophil migration via control of annexin A1 expression. FASEB J. 2017, 31, 3054–3065.
- Ye, R.D.; Boulay, F.; Wang, J.M.; Dahlgren, C.; Gerard, C.; Parmentier, M.; Serhan, C.N.; Murphy, P.M. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the Formyl Peptide Receptor (FPR) Family. Pharmacol. Rev. 2009, 61, 119–161.
- He, H.-Q.; Ye, R.D. The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition. Molecules 2017, 22, 455.
- Babbin, B.A.; Lee, W.Y.; Parkos, C.A.; Winfree, L.M.; Akyildiz, A.; Perretti, M.; Nusrat, A. Annexin I Regulates SKCO-15 Cell Invasion by Signaling through Formyl Peptide Receptors. J. Biol. Chem. 2006, 281, 19588–19599.
- De Jong, R.; Leoni, G.; Drechsler, M.; Soehnlein, O. The advantageous role of annexin A1 in cardiovascular disease. Cell Adhes. Migr. 2017, 11, 261–274.
- Ansari, J.; Kaur, G.; Gavins, F.N.E. Therapeutic Potential of Annexin A1 in Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2018, 19, 1211.
- Fredman, G.; Tabas, I. Boosting Inflammation Resolution in Atherosclerosis: The Next Frontier for Therapy. Am. J. Pathol. 2017, 187, 1211–1221.
- Recio, C.; Maione, F.; Iqbal, A.J.; Mascolo, N.; De Feo, V. The Potential Therapeutic Application of Peptides and Peptidomimetics in Cardiovascular Disease. Front. Pharmacol. 2017, 7, 526.
- Shen, X.; Zhang, S.; Guo, Z.; Xing, D.; Chen, W. The crosstalk of ABCA1 and ANXA1: A potential mechanism for protection against atherosclerosis. Mol. Med. 2020, 26, 1–8.
- Caso, V.M.; Manzo, V.; Pecchillo Cimmino, T.; Conti, V.; Caso, P.; Esposito, G.; Russo, V.; Filippelli, A.; Ammendola, R.; Cattaneo, F. Regulation of Inflammation and Oxidative Stress by Formyl Peptide Receptors in Cardiovascular Disease Progression. Life 2021, 11, 243.
- McKanna, J.A.; Chuncharunee, A.; Munger, K.A.; Breyer, J.A.; Cohen, S.; Harris, R.C. Localization of p35 (annexin I, lipocortin I) in normal adult rat kidney and during recovery from ischemia. J. Cell. Physiol. 1992, 153, 467–476.
- Salles, J.P.; Gayral-Taminh, M.; Fauvel, J.; Delobbe, I.; Mignon-Conté, M.; Conté, J.J.; Chap, H. Sustained effect of angiotensin II on tyrosine phosphorylation of annexin I in glomerular mesangial cells. J. Biol. Chem. 1993, 268, 12805–12811.
- Neymeyer, H.; Labes, R.; Reverte, V.; Saez, F.; Stroh, T.; Dathe, C.; Hohberger, S.; Zeisberg, M.; Müller, G.A.; Salazar, J.; et al. Activation of annexin A1 signalling in renal fibroblasts exerts antifibrotic effects. Acta Physiol. 2015, 215, 144–158.
- Dang, V.D.; Jella, K.K.; Ragheb, R.R.T.; Denslow, N.D.; Alli, A.A. Lipidomic and proteomic analysis of exosomes from mouse cortical collecting duct cells. FASEB J. 2017, 31, 5399–5408.
- Vervoordeldonk, M.J.; Schalkwijk, C.G.; Vishwanath, B.S.; Aarsman, A.J.; van den Bosch, H. Levels and localization of group II phospholipase A2 and annexin I in interleukin- and dexamethasone-treated rat mesangial cells: Evidence against annexin mediation of the dexamethasone-induced inhibition of group II phospholipases A2. Biochim. Biophys. Acta (BBA)-Bioenerg. 1994, 1224, 541–550.
- Seidel, S.; Neymeyer, H.; Kahl, T.; Röschel, T.; Mutig, K.; Flower, R.; Schnermann, J.; Bachmann, S.; Paliege, A. Annexin A1 modulates macula densa function by inhibiting cyclooxygenase 2. Am. J. Physiol.-Renal Physiol. 2012, 303, F845–F854.
- Abboud, H.E. Mesangial cell biology. Exp. Cell Res. 2012, 318, 979–985.
- Bruschi, M.; Sinico, R.A.; Moroni, G.; Pratesi, F.; Migliorini, P.; Galetti, M.; Murtas, C.; Tincani, A.; Madaio, M.; Radice, A.; et al. Glomerular Autoimmune Multicomponents of Human Lupus Nephritis In Vivo: α-Enolase and Annexin AI. J. Am. Soc. Nephrol. 2014, 25, 2483–2498.
- Everts-Graber, J.; Martin, K.R.; Thieblemont, N.; Mocek, J.; Roccabianca, A.; Chafey, P.; Le Gall, M.; Tacnet-Delorme, P.; Reutelingsperger, C.P.; Naccache, J.-M.; et al. Proteomic analysis of neutrophils in ANCA-associated vasculitis reveals a dysregulation in proteinase 3-associated proteins such as Annexin-A1 involved in apoptotic cell clearance. Kidney Int. 2019, 96, 397–408.
- Wu, Y.; Wang, L.; Meng, L.; Cao, G.-K.; Zhang, Y. Evaluation of CRRT effects on pyemic secondary AKI by serum cartilage glycoprotein 39 and Annexin A1. Exp. Ther. Med. 2016, 12, 2997–3001.
- Araujo, L.P.; Truzzi, R.R.; Mendes, G.E.; Luz, M.A.; Burdmann, E.A.; Oliani, S.M. Interaction of the Anti-Inflammatory Annexin A1 Protein and Tacrolimus Immunosuppressant in the Renal Function of Rats. Am. J. Nephrol. 2010, 31, 527–533.
- Araujo, L.P.; Truzzi, R.R.; Mendes, G.E.F.; Luz, M.A.M.; Burdmann, E.A.; Oliani, S.M. Annexin A1 protein attenuates cyclosporine-induced renal hemodynamics changes and macrophage infiltration in rats. Inflamm. Res. 2011, 61, 189–196.
- Ka, S.-M.; Tsai, P.-Y.; Chao, T.-K.; Yang, S.-M.; Hung, Y.-J.; Chen, J.-S.; Shui, H.-A.; Chen, A. Urine Annexin A1 as an Index for Glomerular Injury in Patients. Dis. Markers 2014, 2014, 854163.
- Northcott, C.A.; Glenn, J.P.; Shade, R.E.; Kammerer, C.M.; Hinojosa-Laborde, C.; Fink, G.D.; Haywood, J.R.; Cox, L.A. A custom rat and baboon hypertension gene array to compare experimental models. Exp. Biol. Med. 2012, 237, 99–110.
- Purvis, G.S.D.; Chiazza, F.; Chen, J.; Azevedo-Loiola, R.; Martin, L.; Kusters, D.H.M.; Reutelingsperger, C.; Fountoulakis, N.; Gnudi, L.; Yaqoob, M.M.; et al. Annexin A1 attenuates microvascular complications through restoration of Akt signalling in a murine model of type 1 diabetes. Diabetologia 2018, 61, 482–495.
- Adel, F.W.; Rikhi, A.; Wan, S.-H.; Iyer, S.R.; Chakraborty, H.; McNulty, S.; Tang, W.W.; Felker, G.M.; Givertz, M.M.; Chen, H.H. Annexin A1 is a Potential Novel Biomarker of Congestion in Acute Heart Failure. J. Card. Fail. 2020, 26, 727–732.
- Zimmermann, U.; Woenckhaus, C.; Teller, S.; Venz, S.; Langheinrich, M.; Protzel, C.; Maile, S.; Junker, H.; Giebel, J. Expression of annexin AI in conventional renal cell carcinoma (CRCC) correlates with tumour stage, Fuhrman grade, amount of eosinophilic cells and clinical outcome. Histol. Histopathol. 2007, 22, 527–534.
- Niinivirta, M.; Enblad, G.; Edqvist, P.-H.; Pontén, F.; Dragomir, A.; Ullenhag, G.J. Tumoral ANXA1 Is a Predictive Marker for Sunitinib Treatment of Renal Cancer Patients. J. Cancer 2017, 8, 3975–3983.
- Yamanoi, M.; Yamanoi, K.; Fujii, C.; Fukuda, M.N.; Nakayama, J. Annexin A1 expression is correlated with malignant potential of renal cell carcinoma. Int. J. Urol. 2018, 26, 284–290.
- Sadashiv, R.; Bannur, B.M.; Shetty, P.; Dinesh, U.S.; Vishwanatha, J.K.; Deshpande, S.K.; Bargale, A.; Sarathkumar, E.; Ruikar, K. Comparative expression analysis of phospholipid binding protein annexin a1 in nephrogenesis and kidney cancer. J. Basic Clin. Physiol. Pharmacol. 2019, 31, 20190179.
- Allcock, G.H.; Allegra, M.; Flower, R.J.; Perretti, M. Neutrophil accumulation induced by bacterial lipopolysaccharide: Effects of dexamethasone and annexin 1. Clin. Exp. Immunol. 2001, 123, 62–67.
- Brancaleone, V.; Mitidieri, E.; Flower, R.J.; Cirino, G.; Perretti, M. Annexin A1 Mediates Hydrogen Sulfide Properties in the Control of Inflammation. J. Pharmacol. Exp. Ther. 2014, 351, 96–104.
- Tecklenborg, J.; Clayton, D.; Siebert, S.; Coley, S.M. The role of the immune system in kidney disease. Clin. Exp. Immunol. 2018, 192, 142–150.
- Marshall, S.A.; Qin, C.X.; Jelinic, M.; O’Sullivan, K.; Deo, M.; Walsh, J.; Li, M.; Parry, L.J.; Ritchie, R.H.; Leo, C.H. The Novel Small-molecule Annexin-A1 Mimetic, Compound 17b, Elicits Vasoprotective Actions in Streptozotocin-induced Diabetic Mice. Int. J. Mol. Sci. 2020, 21, 1384.
- Facio, F.N., Jr.; Sena, A.A.; Araújo, L.P.; Mendes, G.E.; de Castro, I.; Luz, M.A.M.; Yu, L.; Oliani, S.M.; Burdmann, E.A. Annexin 1 mimetic peptide protects against renal ischemia/reperfusion injury in rats. J. Mol. Med. 2010, 89, 51–63.
- Suliman, H.; Ma, Q.; Zhang, Z.; Ren, J.; Morris, B.T.; Crowley, S.D.; Ulloa, L.; Privratsky, J.R. Annexin A1 Tripeptide Mimetic Increases Sirtuin-3 and Augments Mitochondrial Function to Limit Ischemic Kidney Injury. Front. Physiol. 2021, 12, 683098.
- Wu, L.; Liu, C.; Chang, D.-Y.; Zhan, R.; Sun, J.; Cui, S.-H.; Eddy, S.; Nair, V.; Tanner, E.; Brosius, F.C.; et al. Annexin A1 alleviates kidney injury by promoting the resolution of inflammation in diabetic nephropathy. Kidney Int. 2021, 100, 107–121.
- Wu, L.; Liu, C.; Chang, D.-Y.; Zhan, R.; Zhao, M.; Lam, S.M.; Shui, G.; Zhao, M.-H.; Zheng, L.; Chen, M. The Attenuation of Diabetic Nephropathy by Annexin A1 via Regulation of Lipid Metabolism through AMPK/PPARα/CPT1b Pathway. Diabetes 2021, 70, 2192–2203.
- Chutipongtanate, S.; Fong-Ngern, K.; Peerapen, P.; Thongboonkerd, V. High Calcium Enhances Calcium Oxalate Crystal Binding Capacity of Renal Tubular Cells via Increased Surface Annexin A1 but Impairs Their Proliferation and Healing. J. Proteome Res. 2012, 11, 3650–3663.
- Peerapen, P.; Thongboonkerd, V. Caffeine prevents kidney stone formation by translocation of apical surface annexin A1 crystal-binding protein into cytoplasm: In vitro evidence. Sci. Rep. 2016, 6, 38536.
- Peerapen, P.; Thongboonkerd, V. Protective Cellular Mechanism of Estrogen against Kidney Stone Formation: A Proteomics Approach and Functional Validation. Proteomics 2019, 19, e1900095.
- Foo, S.L.; Yap, G.; Cui, J.; Lim, L.H.K. Annexin-A1–A Blessing or a Curse in Cancer? Trends Mol. Med. 2019, 25, 315–327.
- Kim, S.B.; Yang, W.S.; Lee, S.O.; Lee, K.P.; Park, J.S.; Na, D.S. Lipocortin-1 Inhibits Proliferation of Cultured Human Mesangial Cells. Nephron 1996, 74, 39–44.
This entry is offline, you can click here to edit this entry!