Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Agriculture, Dairy & Animal Science
Genomic safe harbors (GSHs) provide ideal integration sites for generating transgenic organisms and cells and can be of great benefit in advancing the basic and applied biology of a particular species.
- Pv11 cells
- genomic safe harbor sites
- anhydrobiosis
- transgenesis
1. Introduction
Transgene integration, one of the most commonly used and effective techniques in biological research, can be achieved by either of two approaches: random integration of the gene of interest or site-specific knock-in. Random integration is the simpler method, but it can have unwanted and unpredictable effects on the host cell phenotype, depending on the integration site. In contrast, site-specific knock-in results in more controlled outcomes and has been facilitated by recent advances in genome-editing technologies. Furthermore, in some species and cell lines, several research groups have developed genomic sites called genomic safe harbors (GSHs) that enable transgenes to function as designed without adverse effects on the host [1,2,3,4,5,6]. Transgene integration into such GSHs has promoted fundamental and applied biological research, but identifying GSHs is still challenging, especially in non-model species, owing to insufficient development of the genome database and genetic engineering tools.
GSHs are sites in the genome where new genetic material can be integrated without affecting the host phenotype and where the newly integrated material functions as designed [1]. For example, in basic research, a variety of genetic materials have been integrated into a mouse GSH, the Rosa26 locus, allowing exogenous gene expression or highly targeted gene knockdown experiments [7]. In applied research, GSHs have been used to establish cells that can stably produce proteins of medical interest, for example, therapeutic antibodies [4,8] and species-specific glycosylated proteins [9]. In addition, some researchers have proposed a therapy for genetic diseases using cells that are genetically modified via human GSHs [10]. Therefore, the identification and exploitation of GSHs can significantly advance our understanding of the biology of a particular species as well as leading to the development of new biotechnological applications.
GSHs have usually been identified by a four-step approach [4,11,12]: (i) creation of a transgenic cell pool by random integration; (ii) selection of cells showing stable transgene expression and no/few phenotype changes due to the exogenous gene material; (iii) genome analysis of the selected cells; (iv) confirmation of whether the integration sites are GSHs by a site-specific knock-in method. However, there are a number of difficulties in executing this strategy. Thus, for step (i), suitable genetic engineering tools are needed to create transfectants, and these might not be available for the species of interest. Step (ii) is time-consuming and requires a major effort to monitor the functional and phenotypic stability of the integrated genetic material and the transformant cells, respectively. For step (iii), a genome sequence and database are normally required to identify integration sites, while for step (iv), an appropriate site-specific knock-in method, such as the CRISPR/Cas9 system, must be available for the subject species. Therefore, GSH identification is not a straightforward process, especially in non-model species.
Pv11 is a culturable cell line derived from an insect, the sleeping chironomid P. vanderplanki, which inhabits semi-arid regions in Africa [13]. P. vanderplanki larvae display extreme desiccation tolerance [14], and Pv11 has inherited this ability, such that the cells can be preserved in the dry state at room temperature, while retaining their ability to proliferate once rehydrated [13]. When Pv11 cells are dried, any exogenous protein they contain can be preserved at room temperature for up to 372 days; thus, because of their desiccation tolerance, Pv11 cells potentially have a number of industrial applications, for instance, as water-free storage containers for biomaterials at room temperature [15]. The identification of GSHs in Pv11 cells will provide a major step towards such applications.
To explore the molecular mechanisms underlying the desiccation tolerance of Pv11 cells, we have exploited several gene-manipulation techniques, including a random integration method [16] and a site-specific knock-in method using the CRISPR/Cas9 system [17,18], and have generated several key resources. We have produced a transgenic cell pool, so-called KH cells, by random integration of an AcGFP1 (Aequorea coerulescens GFP)-expressing plasmid [16]. Furthermore, we have a well-annotated genomic database and have developed the CRISPR/Cas9 system in Pv11 cells [19]. Thus, the materials needed to identify GSHs of Pv11 cells are already in place.
2. Cloning Subpopulations with Improved Anhydrobiotic Ability from a KH Cell Pool
To isolate clonal KH cell subpopulations, single-cell sorting was performed (Figure 1A). We acquired two cell lines, B2 and 4C, whose survival rates after rehydration were higher than the original KH cells (Figure 1B). The proliferation rates of B2 and 4C were the same and faster than that of KH cells, respectively, although all KH-derived cells grew more slowly than wild-type Pv11 cells (Figure 1C). Thus, the two cell lines displayed a less-impaired phenotype than the original KH cells.
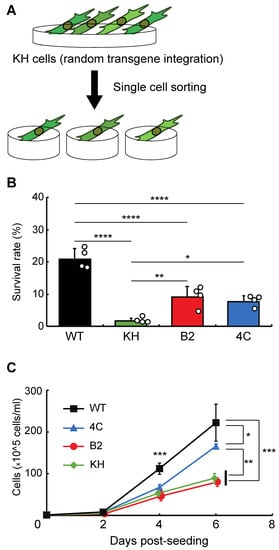
Figure 1. Selection of clonal cell lines from the KH cell pool. (A) The experimental scheme is shown. To establish clonal cell lines from KH cells, single-cell sorting was performed. (B) The survival rates after desiccation–rehydration treatment of the clonal cell lines, B2 and 4C, are shown. (C) The proliferation rates of the B2 and 4C cell lines are shown. Values are expressed as mean ± standard deviation (SD); n = 4 in each group. **** p < 0.0001; *** p < 0.001; ** p < 0.01; * p < 0.05.
3. Genome-Wide Analysis and Identification of the Integration Sites in B2 and 4C Lines
Next, to identify the integration sites in the two cell lines, high-molecular-weight genomic DNA was extracted. DNA libraries were prepared and sequenced with a MinION sequencer. As illustrated in Figure 2A, fragmented plasmid sequences were detected throughout the whole genome (Figure 2B). In contrast, the AcGFP1 expression unit was detected only on chromosome 1 (Chr1; Figure 2B,C) in both clones. Three of these integration sites, Chr1:280397, Chr1:21155382 and Chr1:21164645, were located in intergenic regions, while a fourth, Chr1:21143572, was located in the intron of transcription unit, g12121, whose expression is relatively low in Pv11 cells (Figure 2C, accession number GSE171333 [17]).
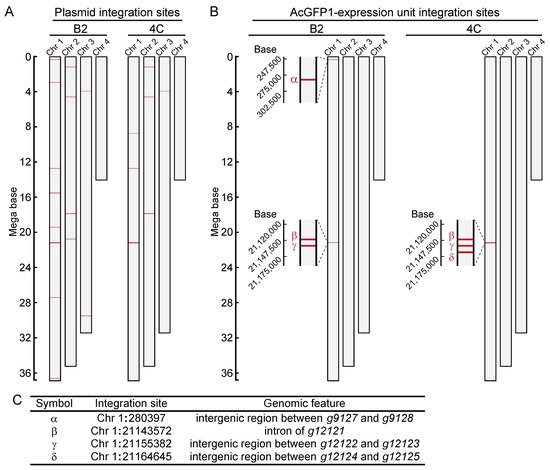
Figure 2. Genome-wide analysis of integration sites of the exogenous plasmid sequence in clonal cell lines, B2 and 4C. (A) Integration sites of the plasmid sequence are shown. (B) Integration sites of the AcGFP1-expression unit are shown. (C) Identified GSH candidates are listed, and their genomic features are described.
4. Identification of Genomic Safe Harbors in Pv11
Next, we examined whether genomic integration at the above sites affects the anhydrobiotic ability or proliferation rate of the corresponding cells. As shown in Figure 3A, AcGFP1 and ZeoR expression units were inserted individually into each genomic site in wild-type Pv11 cells by the CRIS-PITCh method [17,18,22]. Although precise integration was not achieved at Chr1:280397, the other three sites allowed exogenous DNA integration as designed, and this was confirmed by Sanger sequencing. The knock-in cell lines displayed similar desiccation survival rates and proliferation rates to wild-type Pv11 cells (Figure 3B,C). Therefore, the three sites, Chr1:21143572, Chr1:21155382, and Chr1:21164645, were identified as potential GSHs in Pv11.
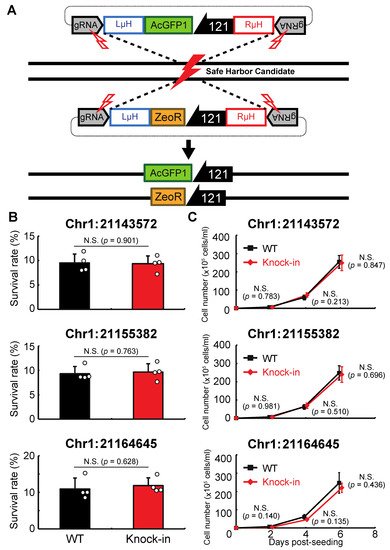
Figure 3. Integration of the expression units of AcGFP1 and zeocin-resistance (ZeoR) genes into GSH candidates. (A) The knock-in scheme for AcGFP1 and ZeoR expression units is shown; the donor vectors harboring AcGFP1 and ZeoR genes under control of the 121 promoter were transfected into Pv11 cells. (B) The survival rate after desiccation–rehydration treatment is shown for each knock-in cell line. (C) The proliferation rate of the knock-in cell lines is shown. Values are expressed as mean ± SD; n = 4 in each group. N.S., not significant.
We then checked the stability of the protein expression level and the cellular phenotypes in a knock-in cell line with a copy of AcGFP1 at Chr1:21164645. The cells were grown for more than one year and, as shown in Figure 4, long-term culture had no effect on AcGFP1 fluorescence intensity (Figure 4A), nor the anhydrobiotic ability (Figure 4B) and proliferation rate (Figure 4C) of the cells.
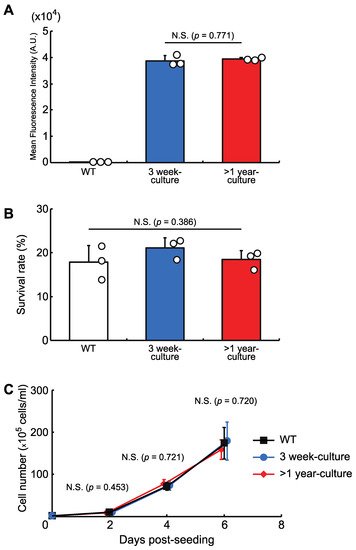
Figure 4. The effects of long-term culture after knock-in at the GSH, Chr1:21164645. (A) AcGFP1-expression stability after more than one year in culture without zeocin. (B) Cell survival rate following desiccation–rehydration treatment after more than one year in culture without zeocin. (C) Cell proliferation rate after more than one year in culture without zeocin. Values are expressed as mean ± SD; n = 3 in each group in (A,B); n = 4 in each group in (C). N.S., not significant.
5. Construction of a GOI Knock-In System for Pv11
Next, we attempted to construct a gene-of-interest (GOI) expression system at the Chr1:21164645 site. A donor vector containing bidirectional HaloTag and AcGFP1 expression unit was designed, the former as an example of a GOI, and the latter as a marker of successful integration (Figure 5A). In our first attempt, a 40 bp homology arm length was used as shown in Figure 3 and described in previous studies [17,18,22]. However, the integration efficiency was low (7.1 ± 1.3% of the target cells were HaloTag+/AcGFP1+ cells; Figure 5B,C), possibly because the insert size was much longer than the homology arm length. Therefore, to determine the optimal homology arm length for this knock-in system, we constructed a series of donor vectors with bidirectional HaloTag and AcGFP1 expression unit flanked by homology arms of various lengths (from 0 to 1000 bp; Figure 5A). Each of these donor vectors was transfected with the previously used construct containing the ZeoR expression unit (Figure 3), and after zeocin selection the protein expression levels of HaloTag and AcGFP1 were analyzed (Figure 5B). There was no significant difference in the knock-in efficiencies of the 250, 500, 750, and 1000 bp HA groups (23.7 ± 2.7%, 26.0 ± 2.6%, 26.0 ± 4.3%, and 25.6 ± 2.9% of cells were HaloTag+/AcGFP1+, respectively), but the 0, 40, and 125 bp HA groups all gave a lower knock-in efficiency (8.8 ± 2.4%, 7.1 ± 1.3%, and 20.7 ± 1.7% of cells were HaloTag+/AcGFP1+, respectively; Figure 5C). Thus, a homology arm length greater than 250 bp is needed for efficient knock-in of the GOI expression construct at the Chr1:21164645 site.
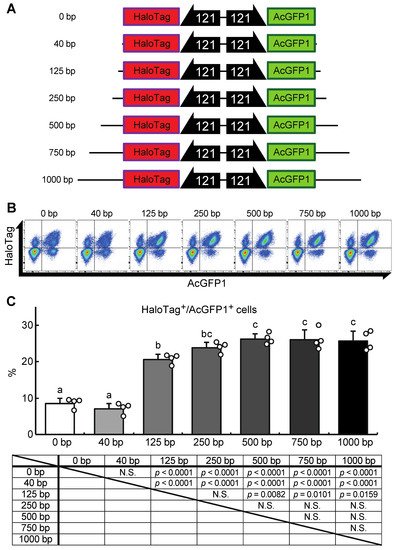
Figure 5. A donor vector construct for GOI expression and optimization of the homology arm length for maximum knock-in efficiency at the Chr1:21164645 site. (A) Schematic outline of donor vectors with different homology arm lengths in the range 0–1000 bp. (B) Representative dot plot data of transfected cells after zeocin selection showing AcGFP1 and HaloTag fluorescence. (C) The proportions of AcGFP1+ and HaloTag+ cells in the live-cell population analyzed using a flow cytometer after 10 days in culture with zeocin selection, and the result of the statistical analysis. Values are expressed as mean ± SD; n = 4 in each group. N.S., not significant. Different letters above each bar indicate significant differences among groups at p = 0.05 as shown in the statistical analysis. The darker shade indicates the longer homology arm length.
To check whether the GOI knock-in construct affects anhydrobiosis and cell proliferation, a clonal HaloTag- and AcGFP1-positive cell line was established, and the genotype and phenotype were analyzed. Sanger sequencing of the genome showed precise integration of the donor vector as designed in Figure 5A, while the cells exhibited the same anhydrobiotic ability and proliferation rate as wild-type Pv11 cells.
This entry is adapted from the peer-reviewed paper 10.3390/genes13030406
This entry is offline, you can click here to edit this entry!