The kidney is one of the most energy-demanding organs and, after the heart, has the second highest expression of proteins involved in mitochondrial function and oxygen consumption. The kidney requires energy mainly for solute reabsorption, among other tasks including waste removal, maintenance of electrolyte and fluid balance and acid–base homeostasis. The generation of an ion gradient across the plasma membrane by Na+/K+-ATPase is essential for solute reabsorption. Therefore, mitochondrial dysfunction is postulated to play a central role in the pathogenesis and progression of kidney diseases including DKD. Mitochondrial dysfunction plays an important role in the pathogenesis and progression of diabetic kidney disease (DKD).
1. Mitochondrial Dysfunction in DKD
Due to the critical role of mitochondria as the powerhouse of cells, mitochondrial dysfunction traditionally referred to an alteration in the production of adenosine triphosphate (ATP) by oxidative phosphorylation (OXPHOS).
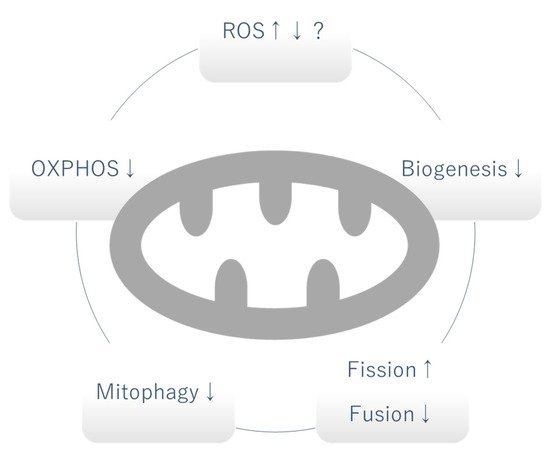
Figure 1. Mitochondrial dysfunction in DKD. Whether the level of mitochondrial ROS is increased or decreased is controversial and can vary depending on the stage of DKD. OXPHOS, mitophagy and biogenesis are generally decreased. Increased fission and decreased fusion causes fragmentation of mitochondria. OXPHOS: oxidative phosphorylation, ROS: reactive oxygen species, ↑: increased, ↓: decreased.
1.1. Mitochondrial Oxidative Phosphorylation (OXPHOS)
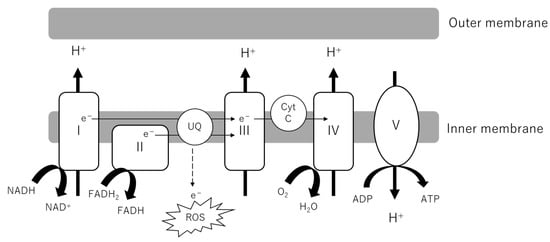
As the powerhouse of cells, the central role of mitochondria is the production of adenosine triphosphate (ATP). Metabolites from glucose, lipids and amino acids are transported into the mitochondrial matrix, serving as substrates of the tricarboxylic acid (TCA) cycle. NADH and FADH
2 are generated along with the reaction feed electrons into Complexes I and II of the electron transport chain (ETC). As electrons are transported through the ETC, H
+ ions are pumped into the intermembrane space. Complex V or ATP synthase uses this proton gradient to generate ATP (
Figure 2). The indices of OXPHOS activity and fitness include oxygen consumption rate (OCR), ATP production, membrane potential and the evaluation of each complex (activity, formation). Generally, it has been observed that OCR in the kidney cortex is increased in early DKD, followed by a decrease as DKD progresses, whereas in glomeruli and podocytes the OCR is decreased in both in early and late phases of the disease [
5]. Although some discrepancy exists between studies, ATP production and complex activity have been demonstrated to be decreased at least in the late stage of DKD [
8,
9]. The contribution of decreased activation of OXPHOS to DKD can be inferred from the observation that some genetic mutations in OXPHOS, such as single-nucleotide polymorphisms (SNPs) in coenzyme Q5 (
COQ5) and cytochrome
c oxidase (
COX6A1), are linked to DKD in humans [
10].
COQ5 encodes methyltransferase located in the mitochondrial matrix and
COX6A1 encodes a subunit of cytochrome c, which is part of the ETC.
Figure 2. Electron transport chain (ETC) in mitochondrial inner membrane. NADH and FADH2 from the TCA cycle donate electrons to Complexes I and II. As electrons are transported through the ETC, a proton gradient is generated, which Complex V or ATP synthase couples to ATP synthesis. Electron leakage from the ETC causes the production of ROS. ADP, adenosine diphosphate; ATP, adenosine triphosphate; Cyt C, cytochrome complex; ROS, reactive oxygen species; UQ, ubiquinone; TCA cycle, tricarboxylic acid cycle.
1.2. Mitochondrial Reactive Oxygen Species (mtROS)
Ever since Brownlee and colleagues proposed that hyperglycemia-induced mitochondrial reactive oxygen species (mtROS) was the unifying mechanism of diabetic microvascular complications in 2000, this paradigm has been prevalent [
11,
12]. Recently, the source of ROS in DKD and the pathogenic role of ROS have become controversial [
13,
14]. Although there may be a consensus that ROS-induced damage is increased in DKD, conflicting studies exist regarding the change in mtROS production, which can be attributed to the different methods used to detect mtROS or the varying models or timepoints of DKD. In both live and fixed db/db mouse kidneys, increased mitochondrial ROS was observed using a mitochondrial matrix-localized reduction–oxidation-sensitive green fluorescent protein probe [
15]. By contrast, in streptozotocin (STZ)-injected C57BL/6J mice and Ins2-Akita mice (DBA/B6 F1 mice), decreased mitochondrial superoxide was observed upon systemic administration of dihydroethidium (DHE) both in live and fixed kidneys [
16]. The latter study does not preclude ROS production in other cell compartments, including the endoplasmic reticulum (ER) or enzyme systems such as nicotinamide adenine dinucleotide phosphate oxidase (Nox). Notably, the restoration of mitochondrial biogenesis and OXPHOS activity by adenosine monophosphate-activated protein kinase (AMPK) activation increased mtROS and ameliorated the DKD phenotype, arguing against the role of mtROS in inciting DKD.
ROS does not play an exclusively detrimental role in cell biology. Mitochondrial hormesis is the concept that slightly enhanced mitochondrial superoxide at baseline can decrease susceptibility to more severe cell stress [
13]. ROS also plays an essential role in certain cell signaling pathways, requiring further elucidation of the intricate characteristics of ROS.
1.3. Mitochondrial Fission and Fusion
Mitochondria are dynamic organelles which undergo tightly controlled processes of fission and fusion. Mitochondrial fission is mediated by dynamin-1-like protein (DRP1) and its receptors such as fission factor 1 (FIS1), mitochondrial fission factor (MFF) and mitochondrial dynamics proteins of 49 and 51 kDa (MID49 and MID51). Mitochondrial fusion is mediated by the long isoforms of optic atrophy protein 1 (OPA1), which plays a role in inner mitochondrial membrane fusion, and the mitofusins (MFN1 and MFN2) which play a role in outer mitochondrial membrane fusion [
5,
6].
Although the increase in mitochondrial fission and fusion factors such as the long isoforms of OPA1, MFN1, MFN2 and MFF were observed in early DKD, mitochondria were consistently fragmented throughout early and late stage in STZ-injected rats [
8]. Human kidney biopsies of patients with DKD also demonstrated fragmented mitochondria in podocytes and proximal tubular cells [
21,
22]. Consistent with increased fission and decreased fusion, Drp1 and FIS1 expression was increased, while MFN2 expression was shown to be decreased in tubules in the latter study.
1.4. Mitophagy
Autophagy is a pathway that degrades and recycles damaged organelles and macromolecules, and selective autophagy of mitochondria is termed as mitophagy. Mitophagy has a critical role in the maintenance of mitochondrial quality by removing damaged mitochondria. Mitophagy can be mediated by the phosphatase and tensin homolog-induced putative kinase 1 (PINK1)/parkin-mediated pathway and other outer mitochondrial membrane proteins such as BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) and NIP3-like protein X (NIX), or the mitophagy receptor FUN14 domain-containing protein 1 (FUNDC1).
The PINK1 and parkin-mediated pathway has been more extensively investigated than the others [
23]. PINK1 has a Ser/Thr kinase domain and is found inserted into both the inner and outer mitochondrial membranes. In healthy mitochondria, PINK1 is cleaved at two points by mitochondrial proteases, leading to its dissociation from the mitochondrial membrane and degradation by the ubiquitin-proteasome system. In depolarized mitochondria, PINK1 escapes cleavage and stably resides in the outer membrane. Subsequently, PINK1 homodimerizes and autophosphorylates to recruit E3 ubiquitin ligase parkin and ubiquitin, directing the mitochondria to the mitophagy pathway. In the ubiquitin-independent pathway, outer mitochondrial membrane proteins such as BNIP3, NIX or FUNDC1 recruit microtubule-associated protein 1A/1B light chain 3 (LC3) and induce mitophagy under certain stimuli including hypoxia [
24,
25]. Cardiolipin, which is located in the inner mitochondrial membrane under normal conditions, is externalized by certain stimuli and detected by LC3, facilitating the engulfment of the mitochondria by autophagosomes [
26]. P62 is a marker of autophagy cargo, and its accumulation can indicate stagnation in degradation via autophagic flux. In general, basal mitophagy levels of podocytes are high, which can be attributed to their terminally differentiated characteristics. In contrast, in tubular cells the mitophagy level is low at baseline but it can be induced as a consequence of stress.
Mitophagy is suppressed in DKD, which was demonstrated by low PINK1/parkin-expression levels in podocytes of STZ-induced diabetic mice and increased p62 expression levels in tubular cells of biopsy obtained from patients with DKD [
27,
28,
29]. Thioredoxin-interacting protein (TXNIP) was implicated in the suppression of tubular autophagy and mitophagy induced by high glucose [
27]. High glucose was also shown to inhibit the transcriptional activity of forkhead-box class O1 (FoxO1) via its phosphorylation by Akt (protein kinase B), leading to the downregulation of PINK1 [
29]. The protective effect of mitoquinone on DKD was partially attributed to the restoration of PINK1 and parkin protein expression in tubular cells via NRF2 activation [
30].
2. Glucose-Induced Mitochondrial Dysfunction in DKD
In glomerular cells, glucose is taken up by glucose transporters (GLUTs) via facilitated diffusion transport. The expression pattern of each member of the GLUTs is cell-specific. Mesangial cells express GLUT1 and 4, podocytes express GLUT1, 4 and 8 and endothelial cells express GLUT1 [
32]. In contrast, tubular cells reabsorb glucose from the glomerular filtrate mainly via sodium-dependent glucose cotransporters (SGLTs). Glucose reabsorbed by tubular cells is dissipated across GLUTs in the basolateral plasma membrane and diffuses into the interstitium. Proximal tubular cells can also produce glucose via gluconeogenesis, which is increased in type 2 diabetes [
33]. Hyperglycemia is the major pathogenetic factor and some intermediate metabolites of glucose metabolism have also been implicated in contributing to cell injury in DKD. In the unifying hypothesis offered by Brownlee and colleagues in 2000, it was proposed that the overproduction of mtROS due to increased influx into OXPHOS activated the nuclear DNA-repair enzyme poly(ADP-ribose) polymerase (PARP), leading to the decreased activity of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and the subsequent accumulation of toxic intermediates of glucose metabolism [
11]. However, later studies have suggested that mitochondria are dysfunctional and that other mechanisms, in addition to a mere increase in substrates, contribute to increased glycolysis and the subsequent accumulation of toxic metabolites.
2.1. Warburg Effect
In general, cancer cells display enhanced glycolysis and impaired oxidative phosphorylation. While the short-time and reversible shift of this metabolic process is called the Crabtree effect, the long-term metabolic reprogramming is called the Warburg effect [
34]. The Warburg effect is the term originally used to describe a shift from OXPHOS to aerobic glycolysis (in which lactate is produced as a final product from glucose) in cancer [
35]. Recent studies have demonstrated that this shift also takes place in diabetic kidneys [
9,
19]. Mitochondria in diabetic tissues are dysfunctional, as discussed above. Transcriptomic, metabolomic and metabolite flux analysis showed increased glucose metabolism and decreased mitochondrial function in the kidney cortices of db/db mice [
9]. A metabolomics analysis of urine samples demonstrated significant decreases of 13 metabolites in patients with DKD compared to healthy controls, 12 of which were associated with mitochondrial metabolism [
19]. Whether mitochondrial dysfunction causes the shift to glycolysis or if increased glycolysis causes mitochondrial dysfunction remains to be established [
36].
Pyruvate kinase is the enzyme which catalyzes the conversion of phosphoenolpyruvate to pyruvate, the last and irreversible step of glycolysis. Decreased pyruvate kinase M2 activity was identified as the possible mechanism of the Warburg effect in DKD by Qi and colleagues [
37]. They first conducted proteomics analysis of glomeruli isolated from patients with type 1 diabetes who did not develop DKD for over 50 years (protected) and those with a histological confirmation of DKD (unprotected). Some enzymes involved in glucose metabolism and antioxidation were found to be increased in protected glomeruli, and in particular, pyruvate kinase M2 (PKM2) expression and activity were upregulated. In mechanistic studies using high-glucose-treated podocytes and STZ-injected mice, it was confirmed that PKM2 activity was decreased in the DKD mouse model, that PKM downregulation contributed to DKD exacerbation and that pharmacological activation of PKM2 reversed the elevation in toxic glucose metabolites and mitochondrial dysfunction. Similarly, decrease in pyruvate kinase activation causes the Warburg effect in cancer [
38].
Other factors that are postulated to induce the Warburg effect in DKD include sphingomyelin and fumarate accumulation [
39]. Matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI) revealed significant increases of ATP/AMP ratio and of a specific sphingomyelin species (SM(d18:1/16:0)) in glomeruli of DKD mice [
40]. In vitro, addition of SM(d18:1/16:0) to mesangial cells activated glycolysis. Fumarate was identified as a factor that mediated Nox4-induced injury in DKD [
41]. Podocyte-specific induction of Nox4 in vivo recapitulated DKD-induced glomerular injury, and metabolomic analysis demonstrated increased fumarate, which was reversed with Nox1/Nox4 inhibition. Fumarate could serve as a hypoxia-inducible factor (HIF) stabilizer, which could cause the activation of glycolysis and suppression of OXPHOS.
2.2. Toxic Metabolites of Glucose Metabolism
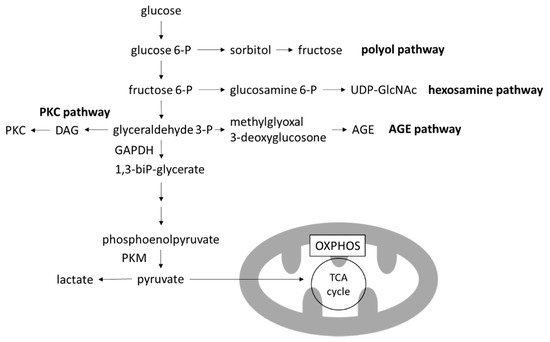
Four major pathways branching from glycolysis are known to produce toxic intermediate metabolites: the polyol pathway, the hexosamine pathway, the advanced glycation end-products (AGEs) pathway and the protein kinase C (PKC) pathway [
12] (
Figure 3). Inhibition of each one of these pathways ameliorates hyperglycemia-induced injury in preclinical models [
42,
43,
44,
45]. Notably, the activation of any of these pathways can be instantly reversed with the restoration of euglycemia.
Figure 3. Glycolysis and branching pathways. In the high-glucose environment of DKD, glycolysis is increased and OXPHOS is decreased, leading to the accumulation of toxic metabolites produced in the branching pathways of glycolysis. AGE, advanced glycation end-product; DAG, diacylglycerol; GAPDH, glygeraldehyde-3-phosphate dehydrogenase; OXPHOS, oxidative phosphorylation; PKC, protein kinase C; PKM, pyruvate kinase M; TCA cycle, tricarboxylic acid cycle; UDP-GlcNAc, uridine diphosphate N-acetylglucosamine.
In particular, levels of 3-deoxyglucosone (3DG) and methylglyoxal (MGO), members of reactive carbonyl species (RCS) which are produced by the degradation of glyceraldehyde in the AGE pathway, are increased with the activation of glycolysis. RCS have highly reactive carbonyl groups and can modify protein and DNA. Proteins glycated by RCS leads to formation of AGEs. Extracellular AGEs can increase the crosslinking of matrices, leading to arterial stiffening [
46]. AGEs can also bind to receptors for AGE (RAGE) and transduce various signals into cells, including nuclear factor-κB (NF-κB) activation leading to ROS formation, inflammation and fibrosis [
47].
This entry is adapted from the peer-reviewed paper 10.3390/biom12030351