Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Engineering, Biomedical
Temperature excursions within a biological milieu can be effectively used to induce drug release from thermosensitive drug-encapsulating nanoparticles. Oncological hyperthermia is of particular interest, as it is proven to synergistically act to arrest tumor growth when combined with optimally-designed smart drug delivery systems (DDSs). Thermoresponsive DDSs aid in making the drugs more bioavailable, enhance the therapeutic index and pharmacokinetic trends, and provide the spatial placement and temporal delivery of the drug into localized anatomical sites.
- hyperthermia
- thermosensitivity
- liposomes
- critical solution temperature
1. Introduction
Nanoparticles-based drug delivery systems (DDSs) combat the adverse limitations associated with conventional treatment regimes, whether in cancer therapy, inflammatory conditions, or cardiovascular diseases such as hypertension and myocardial infarction [1]. Site-specific delivery of therapeutic dosages of the drugs to the diseased cells, while sparing the healthy tissues, is a key advantage of using DDSs. An effective DDS can retain, evade, target, and release its load with controllable and well-regulated kinetics [2].
Nanoparticles (NPs) incorporated in DDSs have been developed over the years, using a wide range of materials, and they exhibit unique chemical and physical properties, allowing them to deliver drugs with high precision. NPs can be broadly classified based on their chemical composition into organic and inorganic. Organic NPs can be lipid-based, such as liposomes and solid lipid NPs, or polymeric-based, such as micelles and dendrimers. On the other hand, inorganic NPs typically contain metals or metal-derivatives in their composition; these include quantum dots (QDs), gold NPs, carbon nanotubes (CNTs), metal organic frameworks (MOFs), and mesoporous silica. While some of these nanoparticles are in the development stage, others have progressed to preclinical and clinical trials. The efficacy of the different nanoparticles as DDSs varies, depending on their size, structure, and physical/chemical properties [3,4,5,6,7]. Such nanocarrier-based systems have especially shown notable implementation in the targeted treatment of cancer, by localizing the effect of antineoplastic agents to diseased tumor sites. Cancer cells induce nearby vascularization (angiogenesis) to meet the increased demands of oxygen and nutrient supply and to sustain rapid proliferation and survival pathways. The resulting intratumoral networks and neovasculature suffer from structural defects in the capillitial endothelium, as well as poor lymphatic drainage and aberrant fluid transport mechanisms [8]. The small size of the NPs allows them to benefit from the innate characteristics of tumors, and they can accumulate at the fenestrae of the tumoral matrices, in a phenomenon known as the enhanced permeability and retention (EPR) effect, which is the basis of passive targeting [9,10].
Besides the merits of passive targeting, active targeting techniques have emerged to further enhance the performance of such DDS. Uncontrolled cell proliferation stimulates the overexpression of certain biomarkers on the cells’ surfaces; nanocarriers can be decorated with motifs or moieties that specifically bind to these receptors. The specificity of active targeting emanates from the complementary arrangement of the overexpressed receptor and the targeting ligand, akin to the lock-and-key mechanism [11]. Besides the specific localization of the nanocarriers, surface functionalization can extend these carriers’ half-life and circulation time in biological systems. For example, PEGylation (modifying the surface with polyethylene glycol chains) has proven to be effective in shielding the nanocarriers from recognition and subsequent rapid elimination from the body by the immune system [12,13].
Conventional NPs generally release their loaded drugs passively, with no control over drug release. While some NPs show quick drug release and undesirable toxicity, the slow drug release from other NPs reduces drug efficiency. Controlling drug release from these NPs, after their accumulation at the targeted site, is essential to ensure spatial and temporal drug release at the diseased site. Advancements in nanomedicine have led to the development of smart NPs that can be triggered to release their load either endogenously (naturally within the body) or exogenously (remotely). Triggers such as ultrasound, light, pH, redox, enzymes, and heat have been widely investigated in the literature as promising stimuli modalities [14,15,16,17,18,19].
2. Thermosensitive Polymers
Heat has been used as a therapeutic tool since the 1800s [20]. In 1866, the German surgeon Carl Busch reported the first account of effective long-term heating for damaging tumor cells without affecting healthy cells [20]. Subsequently, several early in vivo investigations were carried out to examine the thermosensitivity of tumors [21,22,23]. Raising the temperature of the cells to 40–43 °C, for around one hour, can destroy the structure of the cancer cells and affect other cellular processes, making the cells more prone to radiation and antineoplastic drugs [24]. Therefore, hypothermia is used as adjuvant therapy and is usually combined with other cancer treatments, such as radiation, chemotherapy, and immunotherapy [25]. However, it is extremely difficult to produce local hypothermia using the traditional methods without damaging the surrounding healthy tissues, due to prolonged exposure to elevated temperatures. Thermo-sensitive NPs are smart NPs designed to employ the local thermal energy to stimulate a spatial-temporal release of chemotherapeutic drugs at the desired site, while producing no adverse effects on the neighboring healthy tissues of normal temperature. Some NPs, such as liposomes and micro/nanogels, can be designed using a thermo-sensitive composition that changes in structure in response to the increase in the surrounding temperature, leading to a steady release of the encapsulated drug. For example, liposomes can be designed to ‘melt’ (change from the gel state to the liquid state) upon heating to a certain temperature. This type of liposomes is known as the traditional thermosensitive liposomes (TTSL). Thermo-sensitive micro/nanogels, on the other hand, are designed to change their volume by either shrinking or swelling when exposed to a specific temperature, known as the volume phase transition temperature or VPTT. Temperatures lower than the VPTT cause the polymer, forming the microgel, to swell up with the water. Temperatures higher than VPTT will result in causing the polymer to shrink [26,27] and release the encapsulated drug. Thermosensitive micro/nanogels response to the change in temperature is generally through reversible disruption of the hydrogen bonds located between the polymer and the surrounding water [28].
A major advancement in the field of thermoresponsive NPs was the development of different types of polymers that are sensitive to temperature (thermosensitive polymers). Incorporating those polymers within the structure of the NPs allows them to be sensitive or increase their sensitivity to a change in temperature, which alters their structure, resulting in them releasing their load in a controlled manner. NPs incorporating specifically tailored thermosensitive polymers can retain their payload at body temperature (37 °C) but deform and undertake a reversible volume phase transition upon local heating (~40–43 °C) [29].
Potentiating drug release from nanocarrier-based delivery systems using temperature as a triggering modality is a well-established area of research and application. It is essential to fully comprehend the mechanism and rationale behind thermosensitive systems, to develop effective and sophisticated therapeutic platforms. Thermo-responsive nanocarriers incorporate thermosensitive polymers in their structures, which are designed to change their conformation upon exposure to heating/cooling. As the temperature of the surrounding solution increases, thermo-sensitive polymers show coil-to-globule transition. These thermo-responsive polymers usually contain hydrophobic and hydrophilic functionalities, to aid in designing their response. They can transition from the hydration to a dehydration state, in a ‘coil-to-globule’ shift from a homogenously dissolved state to a heterogeneously biphasic state, in response to a small change in temperature (Figure 1) [30]. Previous studies have also shown that both hydrogen bonding and hydrophobic interactions in polymer-solvent systems play a role in the transition from the hydrated random coil to the hydrophobic globule phase, as a result of temperature increase above the critical solution temperatures [31,32]. For example, thermosensitive polymers derived from neutral amphiphilic polymers such as acrylics, carry hydrophilic amide, ether, or alcohol groups and have hydrophobic hydrocarbon backbone chains [33]. Initially, when the homogenous monophasic system exists, the hydrophilic groups in the polymeric network interact readily with water molecules to form hydrogen bonds, resulting in randomly shaped hydrated coils. In response to heating/cooling effects, the conformation is altered where the hydrophilic units become isolated from the aqueous media, such that they are no longer accessible to the water molecules (forming local regions referred to as regions of hydrophobic hydration), initiating crystallization of the polymers to form a biphasic dehydrated nonhomogeneous system [33]. Disruptions to the shape and shrinkage in conformation result in drug release [34].
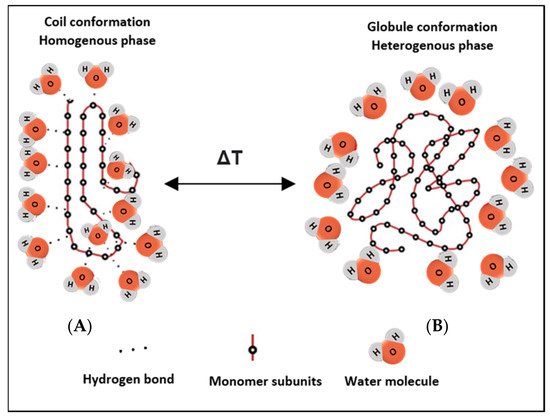
Figure 1. Schematic depicting the reversible phase transition from coil-to-globule and vice versa upon heating/cooling. (A) shows the hydrated state of the polymer, where hydrogen bonds are formed with surrounding water molecules at the hydrophilic ends, while (B) shows the nonhomogeneous state, where the chains dehydrate into globules and fold up, forming a water-rich and a polymer-rich phase. The change in conformation results from a change in the temperature of the system.
An important characteristic of the solutions containing thermoresponsive polymers (polymeric solutions) is their critical solution temperature (CST). Generally, thermoresponsive polymers can be divided into upper critical solution temperature (UCST) and lower critical solution temperature (LCST) types. The phase diagram for each is depicted in Figure 2. Figure 2a shows a UCST system, whereby the polymers would be in the monophasic state above a certain temperate threshold. To alter the conformation of such systems, cooling would be necessary. These are referred to as “positive thermosensitive polymers”. On the other hand, Figure 2b shows an LCST system, and this is the preferred type employed in drug delivery applications. The LCST defines the limiting temperature, above which the system transitions to the binary phase and causes conformation contraction. These polymers are referred to as “negative thermosensitive polymers”. Thermodynamically, a UCST system is an enthalpy-driven system, where interpolymer interactions are more significant and dominant at low temperatures. In contrast, LCST systems are entropy-driven, where an increase in temperature causes the release of the hydrated water molecules (hydrophobic interactions dominate) [34].
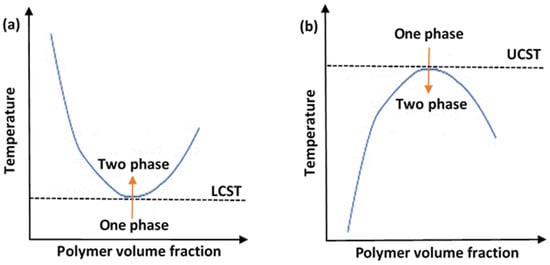
Figure 2. A diagram showing the phase transition behaviors of thermosensitive polymers in aqueous solutions, showing (a) lower critical solution temperature (LCST) system and (b) upper critical solution temperature (UCST) system.
One important LCST polymer is a polyalkylacrylamide derivative, namely poly(N-isopropyl acrylamide) (PNIPAAm). This polymer forms soluble chains in the water below its LCST due to hydrogen bonding between the water and the polymer’s polar groups. Above 32 °C, the waters molecules are expelled from the network, causing the structures to contract, by dehydration of the isopropyl groups [29,30]. The chemical structure of the N-isopropyl acrylamide (NIPAAm) monomer is shown in Figure 3. Generally, the volume phase transition temperature (VPTT) and behavior of thermosensitive hydrogels/microgels can be tailored by changing the balance of the hydrophilic and hydrophobic groups or by introducing an electrostatic charge into the polymer that would influence the polymer–polymer and water–polymer interactions [35]. Thus, copolymerization of PNIPAAm with different monomers that induce different conformational and swelling/de-swelling behaviors from the pure homopolymers conformations is regarded as a flexible strategy to modulate the thermoresponsivity of PNIPAAm-based copolymer systems [35]. The copolymerization monomers can be positively, negatively, or neutrally charged, where philicities, polarities, and concentration come into play. These initiators can be added to the precursor mix during synthesis [35]. Figure 4 shows some of the most common comonomers used to synthesize PNIPAAm copolymers, classified based on charge. For example, the VPTT of PNIPAAm can be changed to ~45 °C by copolymerizing with hydrophilic co-monomers such as acrylamide (AAm), which increase the copolymer chain stiffness and hydrophilicity, as well as limit intramolecular interactions [36]; or with acrylic acid (AAc), which due to its carboxylate groups provides additional repulsive electrostatic interactions that result in a two-step temperature-induced conformational change [37,38]. Polymer size was found to increase with the increase in AAc concentration, as well as a shift in VPTT to a higher temperature due to the hydrophilic nature of AAc [39].
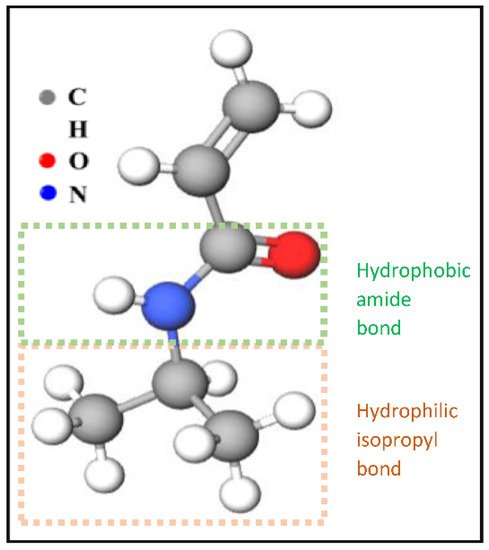
Figure 3. The chemical structure of NIPAAm monomers.
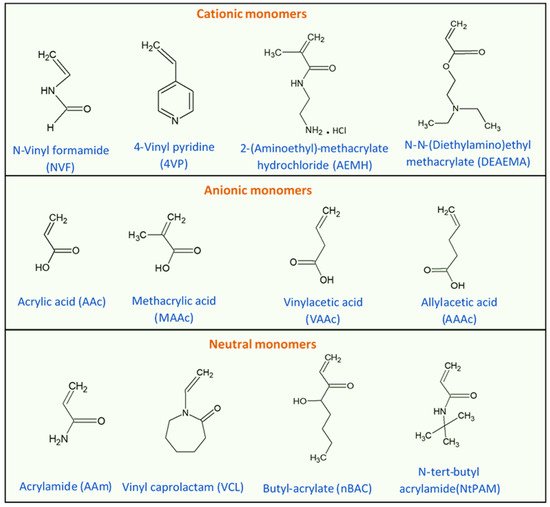
Figure 4. Chemical structures of monomers commonly used in the synthesis of PNIPAAm copolymers with modulated thermoresponsive properties, classified based on their charge.
Although PNIPAAm-based copolymers have garnered great interest in research, there are other thermosensitive particles derived from other types of polymers. For example, Pluronic F127 (Poloxamer 407) is an amphiphilic ABA-type triblock copolymer composed of poly(ethylene oxide)98–poly(propylene oxide)67–poly(ethylene oxide)98 blocks (PEO98–PPO67–PEO98) [40]. It is an attractive biomaterial for the synthesis of thermosensitive drug delivery systems, as it was approved by the FDA for human use. In addition to their reversible gelation capabilities, non-toxicity, biodegradability, and biocompatibility, Pluronic F127-based systems exhibit prolonged drug residence times [40,41]. Table 1 presents some studies that proposed Pluronic F-127-based systems for different drug delivery applications. The main mechanism driving the volume transition above the LCST is attributed to thermal-induced collapse, due to micellization and self-association of the crosslinked Pluronic copolymers dominated by inward hydrophobic interactions [42,43,44]. Such systems with flexible thermal response windows and physical properties have promising potentials for diagnostic and therapeutic applications.
Table 1. Volume change % and transition temperatures (in D2O or PBS) of some thermo-responsive Pluronic F127-based particles.
| Material | Preparation Method | Volume Change (%) | Transition Onset (T °C) | Ref. |
|---|---|---|---|---|
| Pluronic® F127/heparin | Modified emulsification/solvent evaporation method | ~99 | ~25 | [45] |
| Pluronic® F127/poly(ethylenimine) | Modified emulsification/solvent evaporation method | 92–97 | ~21 | [43] |
| Au/Pluronic® F127 | Self-assembly then conjugation | ~96 | ~18 | [44] |
| Pluronic® F127/PEG | Modified emulsification/solvent evaporation method | ~89 | ~23 | [42] |
Thermosensitive nanoparticles (TNPs) benefit from their small size (100–200 nm) in penetrating biological barriers, aiding in the localized delivery of agents and drugs. In addition, the small size of the TNPs allows for rapid reaction to physical changes, as the relaxation time of the volumetric change is directly proportional to the particles’ radius squared (at the critical point) [46]. A rapid transition rate at the critical solution temperature (CST) is always favorable in practical applications. Moreover, TNPs exhibit high specific surface areas compared to particle size, providing a larger number of active sorption sites, which aids in their uptake and biological mobility. Another important property is dispersity of the size distribution, as monodisperse populations exhibit better reaction kinetics to changes at the CST. The current synthesis routes are mostly successful in producing populations with small polydispersity indices, which are calculated as the ratio of weight-average diameter to the number-average diameter of the particles in the distribution [47].
To form thermosensitive polymeric particles with desirable properties, synthesis can be initiated from monomers, polymeric solutions, or macrogels. The most common synthesis routes start with vinyl monomers, which can be neutral or charged. Moreover, a polymeric solution, which contains a crosslinking agent, can be a synthesis precursor or a macrogel that can be physically reduced to form microgel particles [35]. Pelton and Hoare [48] broadly classified the synthesis routes of thermosensitive polymers into three approaches, which are homogenous nucleation, emulsification, and complexation. In the homogenous nucleation approach, the synthesis precursor is a homogenous polymeric solution that contains at least one type of monomer and one crosslinker substance. Including more monomer types increases the complexity of the final product and allows for facile functionalization [35]. The process can be carried out through emulsion polymerization or surfactant free emulsion polymerization (also known as precipitation polymerization). In the emulsion polymerization route, the starting solution contains a suspension of large monomer droplets that are stabilized by surfactant molecules. An example is the preparation of colloidal dispersions of PNIPAAm [49], starting with a precursor solution containing two types of monomers: NIPAAm and methylene-bis-acrylamide, and sodium dodecyl sulfate (SDS) surfactant. The transition temperature of the produced particles was 32 °C, below which the polymer chains would be swollen. Above it, they would shrink due to water rejection, decreasing the average diameter by 2-fold, as a function of the experimental parameters tested. The thermoresponsive behavior and physical properties of the particles was found to be dependent on the concentration of SDS during the polymerization process. Moreover, the particles exhibited a charge due to the carboxyl and sulfate groups from the initiator, which had a noticeable impact on the swelling behavior of the particle at low electrolytic ionic strengths only. This method is widely used because it is robust, versatile, and well-understood. An example of surfactant-free synthesis is the preparation of latex dispersions using monomers of NIPAAm, acrylamide, and N,N′-methylenebisacrylamide [50]. Potassium persulfate was used as an initiator for the free radical polymerization reaction. The precursor solution also contained certain amounts of N,N′-methylenebisacrylamide as a crosslinker. The resulting hydrogels decreased by 10-folds in average diameter upon heating above the LCST. The LCST was found to be a function of acrylamide concentration.
Besides the homogenous nucleation approach, emulsification and complexation are other common routes for thermosensitive particle synthesis. Emulsification is also referred to as inverse emulsion polymerization or mini-emulsion polymerization. A dispersion of hydrophilic monomers in an aqueous phase would be emulsified and polymerized in a continuous non-aqueous phase [35]. Typically, a pre-gel solution (either free monomers or a polymeric solution) is emulsified in oil or any other non-aqueous medium. The droplets are polymerized and crosslinked to form the thermosensitive particles. Polyacrylic acid-based microgels were synthesized using this method by using cyclohexane as the nonpolar continuous medium [51]. Chen et al. [52] characterized the emulsification process of acrylic acid monomers into micelles and concluded that the reaction kinetics (i.e., rate of polymerization) were a direct function of the starting concentration of the monomer. In addition, the monodispersity of the size distribution was dependent on the concentration of the crosslinking agent. The copolymers produced via this synthesis route were robust and less susceptible to coagulation, as the crosslinking agent copolymerizing interfacially yielded hard particles. Meanwhile, complexation depends on forming a colloidal polyelectrolyte complex comprised of two dilute hydrophilic polymers, where one is in excess abundance compared to the other, in order to provide electrostatic stabilization. Feng and colleagues [53] studied the complexation interactions between poly(vinyl amine) and carboxymethyl cellulose and concluded that the mean size of the synthesized complexes was insensitive to the mixing ratio of each polymer, although the particle size distribution was broad. Moreover, mixing the two polymers at different ratios resulted in soluble complexes, colloidal complexes, and macroscopic precipitates; although, stable colloidal complexes were formed only when one of the polymers was provided in excess of the other, in order to contribute to the electrosteric stabilization of the mix. However, the two drawbacks of this approach are (i) the difficulty in separating the excess polymer, and (ii) the high polydispersity index in the particle distribution.
This entry is adapted from the peer-reviewed paper 10.3390/polym14050925
This entry is offline, you can click here to edit this entry!