Epigenetics refers to a scientific domain studying all the processes affecting the expression of genes and/or the activity of transposable elements (TEs) without altering the DNA sequence that may be heritable by mitosis (during development) and/or meiosis (across generations). Forest trees are sessile, perennial, and modular organisms with complex life cycles that are often challenged by environmental variations such as actual climate changes during their long-lifespan. Surviving tree populations can respond to these environmental changes through complex and interacting mechanisms and notably using epigenetics.
- Forest trees
- chromatin
- DNA methylation
- sRNA
- histone modifications and variants
- breeding
- plant biotechnology
- stress
- climate changes
Note:All the information in this draft can be edited by authors. And the entry will be online only after authors edit and submit it.
Definition:The importance of tree genetic variability in the ability of forests to respond and adapt to environmental changes is crucial in forest management and conservation. Along with genetics, recent advances have highlighted “epigenetics” as an emerging and promising field of research for the understanding of tree phenotypic plasticity and adaptive responses. In this entry, we review recent advances in this emerging field and their potential applications for tree researchers and breeders, as well as for forest managers.
1. Epigenetics in Plants
Epigenetics has been initially defined as “the branch of biology which studies the causal interactions between genes and their products, which bring the phenotype into being” [43]. Here, we define it as the study of all the processes affecting the expression of genes and/or the activity of transposable elements (TEs) without altering the DNA sequence that may be heritable by mitosis (during development) and/or meiosis (across generations) [46,47,48] (Figure 1).
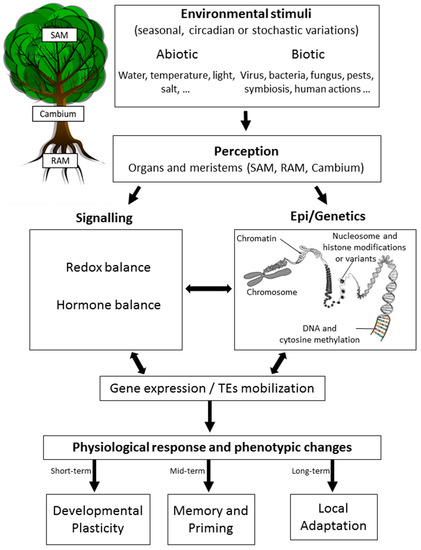
Figure 1. Epigenetic response to environmental changes in trees. Trees are known to recognize various abiotic and/or biotic stimuli occurring rhythmically (circadian or seasonal) or stochastically. These changes are perceived at different tissue levels, with most studies focusing on the leaves, roots, and meristems (SAM, shoot apical meristem; Cambium; or RAM, root apical meristem). Perception is then followed by signaling mechanisms that may include changes in the redox or hormonal balance, which have been shown to be related to epigenetic changes (chromatin remodeling, DNA methylation, non-coding RNA mechanisms (not shown), and histone modifications and variants). This complex crosstalk between signaling processes, epigenetics, and genetics results in an altered gene expression status and/or the mobilization of transposable elements (TEs). A physiological response is then observed together with phenotypic changes that allow trees to acclimate to the environmental changes initially sensed depending on the time-scale considered and the heritable transmission of epigenetic changes.
2. Promises of Epigenetics for Tree Improvement, Breeding, Conservation of Genetic Resources and Forest Management
The potential of epigenetics in plant breeding is already being considered in crops [73,88,89,156,157,158]. Only limited studies have been implemented with trees, and results coming from crops will need to be adapted to fit with the specific features of tree breeding and forest management [40]. Although most of the studies on forest trees have focused on the role of epigenetics in tree development, and response to environmental changes and priming (Table S1), recent studies have pointed out the potential relevance of epigenetics in tree improvement. Three main approaches using epigenetics may be foreseen: (i) Exploit natural or artificially-induced epigenetic diversity; (ii) use epigenetic marks in addition to classic genetic markers for trait improvement with trans-omics approach [159], statistics and modeling, and (iii) use epigenome editing [160]. Epigenetics could be used to expand the material for tree breeders that may be used by sexual or asexual propagation methods.
The best-known example in trees for exploiting natural or induced epigenetic diversity is the existence of an epigenetic memory mechanism that operates during embryo development in Norway spruce (see above section). This process adjusts the timing of bud burst in the progeny, but also in genetically identical epitypes in a manner usually associated with ecotypes in accordance with the temperature conditions during embryogenesis [136,142,143,144,146]. Kvaalen and Johnsen (2008) [143] demonstrated that environmentally induced epigenetic memory during somatic embryogenesis can give similar results for phenology as to those produced by provenance separation of 4–6° latitude. Environmentally induced epigenetic memory during somatic embryogenesis could potentially be used in tree breeding to prime trees regarding their phenology for their first years of plantation along a latitudinal gradient. This promising application opens up many possibilities for forest tree research considering the increasing literature on tree priming (Table S1), and unravels the importance of epigenetic natural diversity that should be taken into consideration for forest breeding, conservation of genetic resources and forest management and protection. Another example of exploiting natural epigenetic variation is emerging from the role of epigenetics in tree heterosis that has been evaluated in Populus deltoides [161]. Findings indicate that methylation patterns of the two P. deltoides parental lines are both partially and dynamically passed on to their intraspecific hybrids, resulting in a non-additive methylation pattern in F1 hybrids. However, studies on epigenetic and heterosis are still necessary for trees and will have to be evaluated by tree breeders.
Another perspective of forest tree epigenetics comes from studies conducted on Arabidopsis and crops concerning sRNAs and cross-kingdom RNA interference (RNAi) [162]. Cai et al. (2018) [163] have shown that Arabidopsis has adapted its exosome-mediated cross-kingdom RNAi as part of its immune responses against pathogens. Pathogens and pests can thus be controlled by sRNAs, targeting their essential or pathogenicity genes, raising the possibility of plants to be protected from diseases by a novel eco-friendly, durable, and highly specific RNA fungicide or pesticide [163,164]. In addition, there is growing evidence to indicate that epigenetic mechanisms directly participate in plant immune responses. However, the evidence of transgenerational inheritance of pathogen-induced defense priming is still a matter of debate [165]. Studies have shown that biotic stresses can also trigger an increase in the overall level of genomic methylation. Curiously, the methylation levels of some pathogen responses or resistance genes are reduced [166]. We still need to understand exactly how epigenetics controls trees’ defenses against disease to then translate this knowledge into practical actions. By exploring natural and induced resistance, we may implement new breeding strategies for preventing disease while reducing the global reliance on harmful pesticides, a field of research still in its infancy for trees.
The application of epigenetic markers to tree breeding has been tested over the last few years, including studies highlighting the extent and variation of genome-wide DNA methylation in natural populations of trees. Ci et al. (2015) [167] investigated the variation in DNA methylation and whether this variation correlates with important plant traits, including leaf shape and photosynthesis in Populus simonii, indicating that epigenetics bridges environmental and genetic factors in affecting plant growth and development. Sow et al. (2018) [125] reported the first estimate of narrow-sense heritability (h2) and phenotypic differentiation (Pst) for global DNA methylation in trees by assessing global DNA methylation variations in Populus nigra clones from natural populations under varying soil water availability. Regardless of water regimes, values of h2 and Pst were comparable to those found for shoot biomass production, a known heritable trait in poplar. Therefore, global DNA methylation, being genetically and environmentally determined in these populations, could be used as a potential marker for population differentiation, performance, and selection under stressful conditions. To explore the epigenetic diversity in breeding programs, we need to expand our knowledge regarding the link between DNA methylation and economic traits in forest trees. Ma et al. (2012) [168] investigated 130 intraspecific hybrids of Populus tomentosa and concluded that the regions defined by the MSAP candidate markers are linked to genes that are essential for photosynthetic traits that respond to DNA methylation which affect growth traits.
The potential of epigenetic markers in quantitative breeding approaches has been recently suggested by Baison et al. (2019) [169]. The authors identified 52 QTLs (Quantitative Trait Loci) of wood properties in Norway spruce for marker-assisted breeding. However, these QTLs explain a small proportion of the genetic variation, in line with previous studies examining genetic variation in complex traits in coniferous species using forward genetic approaches. They suggest that this could be due to several factors, including epigenetic variation, highlighting the need for sophisticated epi/genotyping tools, as well as a combination of advanced statistical models, such as regional heritability mapping. Recently, Champigny et al. (2019) [170] applied statistical learning experiments to genetically diverse populations of Populus balsamifera trees grown at two common garden sites and showed that traits in novel genotypes can be modelled using small numbers of methylated DNA predictors. Indeed, significant phenotypic variances in quantitative traits of the wood were explained by the natural variation of DNA methylation, such as total biomass (57.5%), wood density (40.9%), soluble lignin (25.3%), and carbohydrate content in the cell wall (mannose: 44.8%). The authors proposed that DNA methylation-based models can be used as a strategy to validate the identity, provenance, or quality of agroforestry products. While this approach presents innovative perspectives for forest trees, it should be evaluated in terms of cost, technical, and analytical efforts.
The last approach to use epigenetics for tree improvement is epigenome editing, i.e., a type of genetic engineering in which the epigenome is modified at specific sites using engineered molecules targeted to those sites. Genome editing in trees has been recently reviewed by Bewg et al. (2018) [160], suggesting a great potential for stable CRISPR-induced mutations and associated phenotypes over multiple clonal generations. Authors suggest that this technology can be used for the commercial production of elite trees that relies on vegetative propagation. The potential of epigenome editing to control recombination in plant breeding has been recently reviewed [171]. Manipulating the rate and positions of crossovers to increase the genetic variation accessible to breeders is of main interest. Epi/genome editing at desired sites of recombination, and manipulation of crossovers factors, are applicable approaches for achieving this goal, reducing the time and expense associated with traditional breeding, revealing inaccessible genetic diversity, and increasing control over the inheritance of preferred haplotypes [171]. However, there is no study on epigenome editing in trees, and the feasibility of such technological advances, depending on use regulations in different countries, will have to be evaluated by tree breeders in the future.
Supplementary Materials: The following are available online at https://www.mdpi.com/1999-4907/11/9/976/s1. Table S1: Current state of epigenetics in trees for development, abiotic stress or priming, biotic stress or priming, and markers, breeding and biotechnology topics.
This entry is adapted from the peer-reviewed paper 10.3390/f11090976