Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Amyotrophic lateral sclerosis (ALS) is a fatal disease characterized by the degeneration of cortical and spinal motor neurons. With no effective treatment available to date, patients face progressive paralysis and eventually succumb to the disease due to respiratory failure within only a few years. Intriguingly, a key feature present in both ALS patients and rodent models of the disease is cortical hyperexcitability and hyperconnectivity, the mechanisms of which still remain incompletely understood.
- amyotrophic lateral sclerosis
- excitability
- upper motor neurons
- neurodegeneration
- motor cortex
- microcircuit
- interneurons
- astrocytes
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a lethal disease that is primarily characterized by the loss of upper motor neurons (UMN) in the cortex and lower motor neurons (LMN) in the spinal cord [1][2]. Being one of the most common forms of adult motor neuron diseases, ALS occurs at an incidence of ~2/100,000 person per year and a prevalence of ~6/100,000 people [3]. An ever-growing list of genes and gene variants is linked to the development of ALS [4], but only a mere 5–10% of all cases are familial (fALS) due to inherited genetic mutations, while the large majority of cases (90–95%) are sporadic (sALS) [5][6][7]. Both forms of the disease progress rapidly. The initial muscle weakness, spasticity, and fasciculations soon worsen to cause severe paralysis and eventual fatal respiratory failure, typically within 3–5 years [8]. The mean age of onset of ALS is ~60 years, while in fALS the disease onset already occurs about 5 years earlier [9]. To date, only 40–60% of the familial cases could be linked to known mutations, such as the hexanucleotide repeat expansion in the Chromosome 9 open reading frame 72 (C9orf72, 40% of fALS cases) or mutations in the Superoxide dismutase 1 (SOD1, 20%), the TAR DNA-binding protein (TARDBP, 4%), or in the Fused in sarcoma (FUS, 3%) gene [7][10].
A common pathophysiological feature of ALS is the intracellular aggregation of proteinaceous deposits. The most frequently found (97% of all ALS cases) cytoplasmic aggregates in fALS and sALS consist of the TAR DNA-binding protein of 43 kDa (TDP-43) [11][12]. C9orf72 mutation carriers also display dipeptide repeat proteins [13]. Less frequent are aggregates of FUS, although the actual occurrence rate is a matter of debate [14][15], or SOD1 [16][17]. Genetic insight derived from fALS cases has enabled researchers to study the molecular mechanisms of the disease using animal and in vitro cell culture models. Although the complex molecular changes accompanying motor neuron loss remain to be fully elucidated, these studies have unearthed a plethora of compromised intracellular processes and pathways affecting motor neuron health, involving defective RNA processing [18], impaired oxidative stress regulation and mitochondrial function [19][20], irregularities in cytoskeletal structure affecting axonal transport [21][22], reduced intracellular Ca2+ handling capacity [23][24][25], or compromised energy metabolism [26]. None of which, however, has yet resulted in the development of an effective treatment strategy. ALS research in the past decades was strongly centered on the investigation of lower motor neurons in the spinal cord, while upper motor neuron pathology is understudied. A large body of evidence, however, emphasizes the cardinal role of the motor cortex in triggering UMN and LMN degeneration. As such, cortical hyperexcitability has been demonstrated in ALS patients and even asymptomatic ALS mutation carriers [27], but the molecular, cellular, and network mechanisms underlying cortical hyperexcitability are not completely understood. Both cell autonomous (e.g., altered intrinsic excitability of UMN) and non-cell autonomous mechanisms (e.g., alterations in the local microcircuitry caused by cells other than UMN, such as glia) are conceivable (for detailed review, see Gunes et al. [28]).
2. Clinical Evidence for Cortical Hyperexcitability in ALS
The site of origin of the disease has long been a matter of debate. While some believe the initial pathology occurs at the level of lower motor neurons (LMN) in the spinal cord or even the neuro-muscular junction [29][30], with the cortex and UMN being affected in a retrograde process (termed ‘dying-back hypothesis’), others argue that the disease originates in the cortex and propagates in a corticofugal manner to subsequently affect LMN in the spinal cord [30][31]. The latter process was coined the ‘dying-forward’ hypothesis [31]. Several lines of evidence in fact strongly argue for a cortical origin of ALS [32][33]. These encompass the spreading pattern of the typical TDP-43 pathology, following anatomical connections through axonal projections and synaptic contacts [32]. Most importantly, electrophysiological measurements have revealed that cortical hyperexcitability is present in both sporadic and familial ALS patients and can occur even prior to disease onset in familial ALS cases [34][35][36]. Cortical hyperexcitability can be clinically assessed by means of threshold tracking paired-pulse transcranial magnetic stimulation (TMS) combined with motor-evoked potential (MEP) measurements (e.g., of the M. abductor pollicis brevis), as well as via electroencephalography (EEG) or functional MRI (fMRI) [37][38][39][40][41][42].
A number of parameters can be assessed to probe differences in cortical excitability when performing TMS, such as the resting motor threshold (RMT) and the MEP amplitude [43]. RMT represents the minimal stimulus intensity needed to excite the corresponding muscle [44] and was found to be decreased early in the disease, thereby reflecting UMN hyperexcitability [45][46]. In late-stage ALS patients, however, the RMT was increased along with a decrease in the MEP amplitude, indicative of gross denervation [47][48]. Cortical hyperexcitability could in principle result from either increased excitation or compromised intracortical inhibition. To disentangle both options, paired-pulse stimulation protocols can be applied, in which the interstimulus interval (ISI) determines the magnitude of the MEP. While very short ISIs (1–7 ms) depress the MEP, longer ISI (10–30 ms) can facilitate the MEP [36]. The degree of inhibition or facilitation can be assessed by altering the stimulus intensity of the 2nd pulse, the difference of which is represented in the short/long intracortical inhibition (SICI/LICI) and the intracortical facilitation (ICF) metric [36][38] (Figure 1a–c). In ALS patients, the SICI was shown to be reduced [36][49][50][51][52][53][54][55] (Figure 1b), also in patients with subclinical UMN damage [56], indicating compromised GABAB receptor mediated intracortical inhibition [57]. On the other hand, cortical facilitation was strongly increased, arguing for an elevated excitation [36][58][59][60][61] (Figure 1c). The phenomena of cortical hyperexcitability thus can be explained by a combined increase in excitation and impaired inhibition.
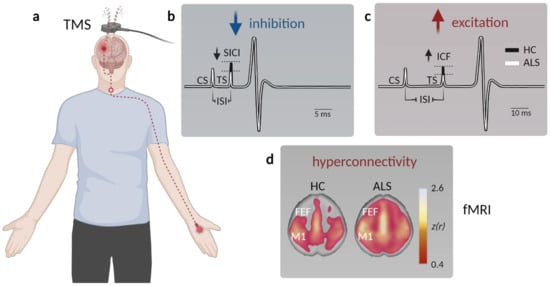
Figure 1. Assessment of motor cortex hyperexcitability and hyperconnectivity in ALS patients. (a) Schematic displaying paired-pulse transcranial magnetic stimulation (TMS) in patients. A magnetic coil is placed above the primary motor cortex (M1), and the resulting motor-evoked potential (MEP) is measured at the innervated muscle (typically the M. abductor pollicis brevis). (b) Example MEP depicting muscle potential changes (black—healthy controls, HC; white—ALS) triggered by a short interstimulus interval (ISI) between the conditioned stimulus (CS) and the test stimulus (TS) to facilitate short-interval intracortical inhibition (SICI). The TS needed to evoke a similar MEP is reduced in ALS, indicative of compromised intracortical inhibition. (c) To probe intracortical facilitation (ICF), the TS is applied after a longer ISI, which typically elicits a stronger MEP response compared to a single stimulus. The TS intensity needed to evoke a similar MEP observed after pair-pulse stimulation in healthy controls is reduced in ALS patients, arguing for elevated excitation within M1. (d) Functional magnetic resonance imaging (fMRI) studies demonstrate enhanced connectivity in somatosensory networks, as well as across (pre)frontal areas (FEF, frontal eye field) in ALS patients (data from Schulthess et al. [27], modified and used with permission of the authors and the publisher).
In addition to TMS, EEG is widely used to assess cortical excitability by measuring parameters such as event-related desynchronization/synchronization (ERD/ERS), enabling higher temporal resolution compared to TMS. ERD typically represents a correlate of an activated cortical network prior to the execution of a motor task [62], whereas ERS indicates an active inhibitory process of the cortical network after the motor task is performed [62]. In support of the notion of cortical hyperexcitability, ALS patients exhibit lower ERS, whereas task-dependent ERD is either decreased—likely reflecting UMN loss during later disease stages—or not significantly altered between ALS-patients and healthy controls [63][64]. These findings can be explained by both increased excitability of UMN and compromised intracortical inhibition during rest and motor task execution [46][61][65]. Corroborating EEG findings, a magnetoencephalography (MEG) study by Proudfoot et al. showed that ALS patients have an enhanced cortical beta desynchronization during a visually cued lateralized motor task [66].
Although limited in number, functional magnetic resonance imaging (fMRI) investigations were also employed in clinical ALS studies. fMRI is a widely used imaging technique that detects changes in blood oxygenation in the brain as a proxy for neuronal activity in the respective region [67][68]. Multiple studies reported a stronger response to a motor task in ALS patients compared to healthy subjects [69][70][71]. Resting state fMRI also revealed an increased functional connectivity of the motor cortex not only within the sensorimotor network [72][73] and the prefrontal association cortex [74], but also within the brainstem, ventral attention, and default mode network in sALS patients [27][75] (Figure 1d). Interestingly, changes in functional connectivity between the cerebellum and a network comprised of the precuneus, the cingulate, and the middle frontal lobe were already observed in presymptomatic mutation carriers [39].
The electrophysiological findings of compromised intracortical inhibition were further substantiated by proton magnetic resonance spectroscopy (1H-MRS) measurements, revealing reduced GABA levels in ALS patients [76] and demonstrated a reduction of glutamate-glutamine levels upon riluzole treatment [77]. Additional histological and transcriptomic analyses demonstrated decreased GABAA receptor densities and a downregulation of the α1-subunit and upregulation of β1-subunit mRNA levels in the post-mortem motor cortex of ALS patients [78]. Furthermore, a downregulation of N-methyl-D-aspartate receptors (NMDAR) and alterations in the expression of particular α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) subunits in ALS were revealed by immunohistochemistry and RT-PCR [79]. These investigations, however, were conducted in post-mortem tissue, and thus reflect end-stage alterations.
Taken together, electrophysiological and imaging studies have revealed an early increase in cortical excitability in ALS, which is likely based on a combined increase in excitation and a decrease in inhibition.
3. Circuit Mechanisms Involved in Cortical Hyperexcitability
The M1 microcircuit consists of excitatory pyramidal neurons (PN) and local inhibitory interneurons (Figure 2a). Depending on their projection target, PN can be further categorized into intratelencephalic (IT) and pyramidal tract neurons (PT, aka UMN, also known as corticospinal motor neurons (CSMN) or corticospinal tract (CST) neurons and Betz cells in humans). IT neurons are found in layer 2/3, 5, and 6, with corticocortical (CC) neurons projecting transcallosally and corticostriatal (CStr) neurons projecting to the striatum. UMN are embedded in layer 5B of M1 (Figure 2a). They are multi-projectional, and downstream target regions include both the ipsi- and contra-lateral brainstem and the spinal cord, with branches also innervating the ipsilateral cortex and some subcortical regions [80]. The strongest feedforward excitatory drive to layer 5 neurons is provided by layer 2/3 PN (Figure 2a), followed by intralaminar projections within layer 5. Activity levels and information processing in M1 is regulated by a diverse set of local and remote inhibitory neurons, which constitute ~20% of the cortical neuronal population [81][82]. Based on the expression of typical markers, three major non-overlapping subtypes of interneurons are known: parvalbumin (PV), somatostatin (SST), and 5-HT3a (with the majority of them being vasoactive intestinal peptide (VIP)) expressing interneurons [83][84][85] (Figure 2a). While PV and SST target PN directly at the level of their somata (PV) and the dendritic compartment (SST), respectively, VIP regulate the circuit via disinhibition, i.e., by inhibiting PV and SST. Notably, SST are also inhibiting PV to allow for further fine-tuning of information processing within neural circuits [86][87]. Glutamatergic (excitatory) and GABAergic (inhibitory) neurons in the M1 circuitry are further regulated by neuromodulatory inputs [80][88][89][90]. Several circuit elements were shown to be structurally and functionally altered in various models of the disease (Figure 2b, Gunes et al. [28]), causing a disruption of the tightly controlled excitation/inhibition balance in M1:
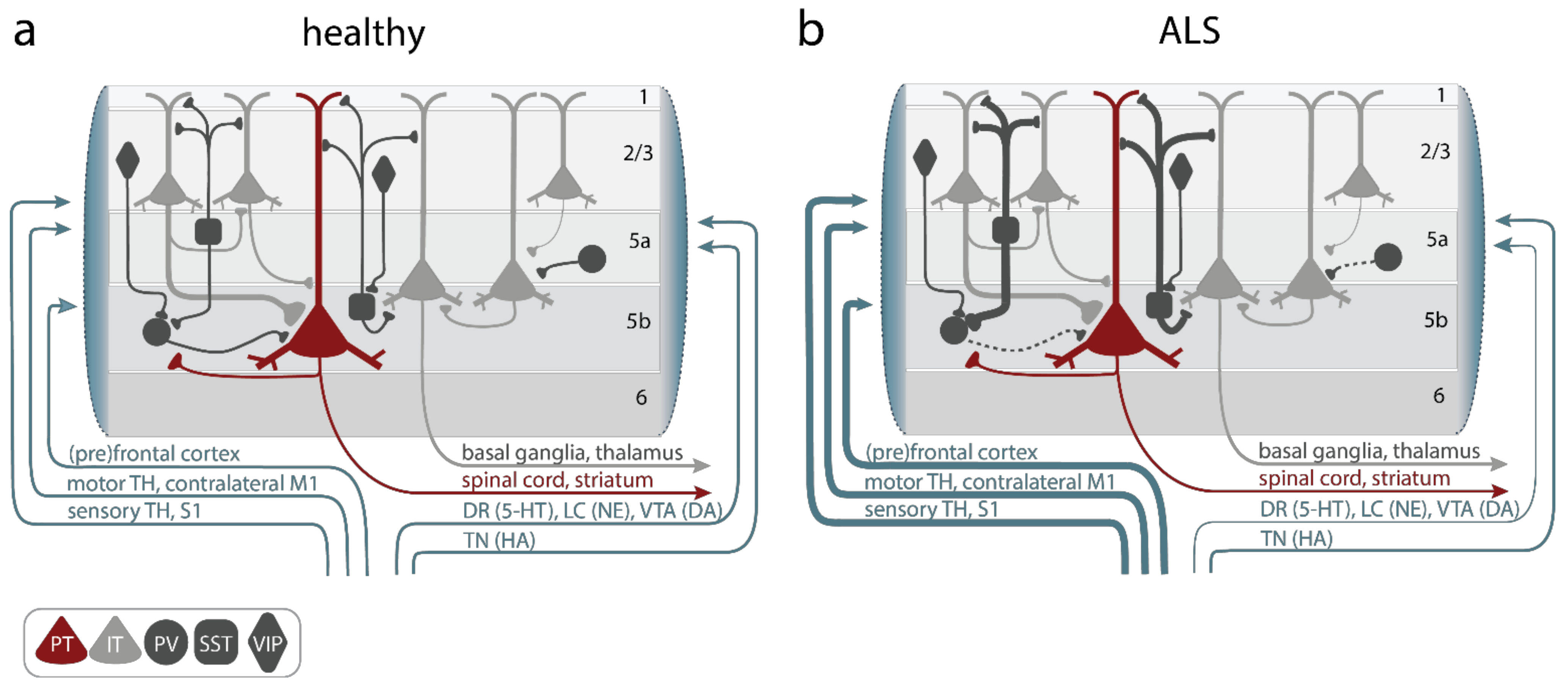
Figure 2. Alterations of primary motor cortex (M1) microcircuit elements in ALS. (a) In the healthy M1 microcircuitry upper motor neurons (UMNs), aka, pyramidal tract neurons (PT, red), located within cortical layer 5b receive feedforward excitatory synaptic input primarily from intratelencephalic (IT, light gray) neurons in layer 2–3 and, to a lesser degree, intralaminarly from layer 5 IT. Long-range feedforward input is provided by the (pre)frontal and somatosensory cortex, the contralateral primary motor cortex (M1), and the thalamus (TH). Excitation is controlled by a number of inhibitory GABAergic interneurons, which express either parvalbumin (PV), somatostatin (SST), or vasoactive intestine peptide (VIP) (dark gray). Neuromodulatory input further shapes information processing in M1 and is provided by projections from the locus coeruleus (LC; releasing norepinephrine (NE)), the ventral tegmental area (VTA; releasing dopamine (DA)), the dorsal raphe (DR; releasing serotonin (5-HT)), or the tuberomammillary nucleus (TN; releasing histamine (HA)). (b) In ALS, hypoactive PV and hyperactive SST interneurons were reported, as well as increased long-range feedforward excitatory synaptic inputs and elevated functional connectivity, while neuromodulatory inputs were found to be compromised. Line thickness reflects connectivity strength.
3.1. Alterations of Upper Motor Neurons
Naturally, UMN have been the center of attention when it comes to studying M1 pathology in ALS. These studies revealed structural abnormalities, which, however, seemingly differ between mouse models: While, in SOD1G93A transgenic (tg) mice, a reduction of dendritic spine density (the structural correlates of excitatory synapses) along with a reduced cell complexity (fewer dendrites) were observed [91][92], an increase in apical and basal dendritic spine densities in TDP43Q331K tg mice was found [93]. Functionally, UMNs were shown to be hyperexcitable across various ALS mouse models. In the SOD1G93A mouse model layer, 5 PNs (which largely comprise UMN) display age- and disease-stage specific alterations in their intrinsic properties. Already in neonatal tg mice (P5–6), UMNs are hyperexcitable, followed by a normalization during pre-symptomatic stages (P14–P70), but there is a reoccurrence of hyperexcitability during symptomatic stages (≥P90) [94][95]. However, in contrast others also reported UMN hypoexcitability during the early symptomatic phase [96]. Similar findings were made in the TDP-43A315T mouse model, in which layer 5 PN displayed a higher firing frequency during the pre-symptomatic stage [2][93]. In agreement with these studies, 30 days of induced expression of TDP-43ΔNLS in adult mice also lead to intrinsic hyperexcitability in M1 layer 5 PN, highlighting the role of TDP-43 cytoplasmic mislocalization in driving cortical hyperexcitability [97]. Furthermore, also in the wobbler mouse model of ALS, which is based on a spontaneous mutation in the VPS54 gene in the C57BL/Fa strain [98], layer 5 PN are hyperexcitable, as shown by an increased input resistance and strongly reduced current threshold (the minimal injected current needed to elicit an action potential) [99].
3.2. Increased Excitatory Inputs to UMNs
In addition to intrinsic alterations of UMNs, there is also evidence for an augmented synaptic input onto them, witnessed as increased miniature excitatory synaptic current (mEPSC) frequencies in UMNs in presymptomatic SOD1G93A [92] and in TDP-43Q331K tg mice [93]. The source of these excitatory synaptic inputs remains elusive. Based on the microcircuit connectivity, the main input source for layer 5 PN (UMNs) are layer 2/3 PN. Notably, layer 2/3 PN are also hyperexcitable in the SOD1G93A model [94] and more active in the FUSΔNLS mouse model, as witnessed by increased spontaneous activity [100], further supporting the notion of excess excitatory drive onto UMNs. Additional excess synaptic inputs might also result from long-range projections. Indeed, connectivity changes were reported in SOD1G93A mice [74], with increased synaptic inputs from S1 and contralateral M2 already during early-symptomatic stages, which further expanded to include the thalamus, contralateral M1, auditory cortex, and caudoputamen at later stages of the disease (Figure 2b, Commisso et al. [74].
3.3. Reduced Cortical Inhibition
Excitation/inhibition (E/I) imbalance can be caused by increased excitation as well as compromised inhibition. In addition to enhanced intrinsic excitability and excitatory inputs onto UMNs, several lines of evidence highlight a parallel reduction in synaptic inhibition in M1 in various mouse models of the disease. Impaired inhibition was evident by reduced inhibitory postsynaptic currents (IPSCs) in UMNs in SOD1G93A [101], TDP-43A315T mice [2] and in the wobbler mouse model [99], which could be based on a loss of interneurons or particularly inhibitory synapses and/or functional interneuronal deficits. Indeed, the researchers recent work in the FUSΔNLS model suggests that primarily inhibitory synapses are affected and less abundant, while overall PV interneuron number was not significantly altered [100]. There are contradictory findings regarding the density of subtype-specific interneurons in SOD1G93A mice. While some have reported no change in the density of PV-, SST-, or VIP- expressing interneurons [102][103], others observed a decrease [101]. In the wobbler mouse model, the density of PV and SST interneurons was decreased [99]. Functionally, mouse-line and disease-stage specific alterations of interneuron subtypes were also identified. In SOD1G93A mice, whole-cell recordings of PV interneurons revealed variable changes throughout the course of the disease [94]. While the excitability of PV interneurons was not altered presymptomatically, they turned hyperexcitable during the symptomatic stage (P90–101) [94]. Others, however, have reported hypoexcitability of PV interneurons in presymptomatic SOD1G93A mice [101] (Figure 2b). Notably, in TDP-43A315T mice Zhang et al. observed hyperactive SST, which suppressed PV activity, thus causing layer 5 PN disinhibition [2] (Figure 2b). Importantly, means to restore PV activity levels in the motor cortex either by ablating hyperactive SST in TDP-43A315T mice [2] or through direct chemogenetic stimulation of PV in SOD1G93A mice [101] restored layer 5 PN excitability and firing rates [2][101], further emphasizing the role of interneurons in cortical ALS pathophysiology.
3.4. Altered Neuromodulation
In addition to glutamatergic and GABAergic inputs, neuronal activity levels are furthermore regulated by a number of neuromodulators, which also seem to be altered in ALS patients and mouse models of the disease [104]. These alterations affect the dopaminergic system, as evidenced by compromised dopaminergic signaling in the striatum in ALS patients [105][106], a finding that is corroborated by mouse studies, in which SOD1G1H mice display a reduction of dopamine levels and the loss of dopaminergic neurons (Figure 2b, Kostic et al. [107]). Moreover, serotonin levels were also found to be affected. In ALS patients, a loss of serotonergic neurons in the brainstem was reported, and also in SOD1G86R mice reduced levels of serotonin in the brainstem and spinal cord were observed [108].
This entry is adapted from the peer-reviewed paper 10.3390/ctn6010005
References
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955.
- Zhang, W.; Zhang, L.; Liang, B.; Schroeder, D.; Zhang, Z.-W.; Cox, G.A.; Li, Y.; Lin, D.-T. Hyperactive somatostatin interneurons contribute to excitotoxicity in neurodegenerative disorders. Nat. Neurosci. 2016, 19, 557–559.
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071.
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102.
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172.
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28.
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310.
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206.
- Mehta, P.R.; Jones, A.R.; Opie-Martin, S.; Shatunov, A.; Iacoangeli, A.; Al Khleifat, A.; Smith, B.N.; Topp, S.; Morrison, K.E.; Shaw, P.J.; et al. Younger age of onset in familial amyotrophic lateral sclerosis is a result of pathogenic gene variants, rather than ascertainment bias. J. Neurol. Neurosurg. Psychiatry 2018, 90, 268–271.
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2013, 17, 17–23.
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438.
- Neumann, M.; Sampathu Deepak, M.; Kwong Linda, K.; Truax Adam, C.; Micsenyi Matthew, C.; Chou Thomas, T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark Christopher, M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133.
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 2013, 339, 1335–1338.
- Deng, H.-X.; Zhai, H.; Bigio, E.H.; Yan, J.; Fecto, F.; Ajroud, K.; Mishra, M.; Ajroud-Driss, S.; Heller, S.; Sufit, R.; et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 2010, 67, 739–748.
- Ikenaka, K.; Ishigaki, S.; Iguchi, Y.; Kawai, K.; Fujioka, Y.; Yokoi, S.; Abdelhamid, R.F.; Nagano, S.; Mochizuki, H.; Katsuno, M.; et al. Characteristic Features of FUS Inclusions in Spinal Motor Neurons of Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2020, 79, 370–377.
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2020, 92, 86–95.
- Benson, B.C.; Shaw, P.J.; Azzouz, M.; Highley, J.R.; Hautbergue, G.M. Proteinopathies as Hallmarks of Impaired Gene Expression, Proteostasis and Mitochondrial Function in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2021, 15, 783624.
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Bennett, C.F.; Cleveland, D.W.; Yeo, G.W. Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res. 2012, 1462, 3–15.
- Kawamata, H.; Manfredi, G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech. Ageing Dev. 2010, 131, 517–526.
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641.
- Xiao, S.; McLean, J.; Robertson, J. Neuronal intermediate filaments and ALS: A new look at an old question. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2006, 1762, 1001–1012.
- Marinković, P.; Reuter, M.S.; Brill, M.S.; Godinho, L.; Kerschensteiner, M.; Misgeld, T. Axonal transport deficits and degeneration can evolve independently in mouse models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 4296–4301.
- Leal, S.S.; Cardoso, I.; Valentine, J.S.; Gomes, C.M. Calcium Ions Promote Superoxide Dismutase 1 (SOD1) Aggregation into Non-fibrillar Amyloid: A Link to Toxic Effects of Calcium Overload in Amyotrophic Lateral Sclerosis (ALS)? J. Biol. Chem. 2013, 288, 25219–25228.
- Lautenschlaeger, J.; Prell, T.; Grosskreutz, J. Endoplasmic reticulum stress and the ER mitochondria calcium cycle in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 166–177.
- Grosskreutz, J.; Bosch, L.V.D.; Keller, B.U. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 2010, 47, 165–174.
- Dupuis, L.; Pradat, P.-F.; Ludolph, A.C.; Loeffler, J.-P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011, 10, 75–82.
- Schulthess, I.; Gorges, M.; Müller, H.-P.; Lulé, D.; Del Tredici, K.; Ludolph, A.C.; Kassubek, J. Functional connectivity changes resemble patterns of pTDP-43 pathology in amyotrophic lateral sclerosis. Sci. Rep. 2016, 6, 38391.
- Gunes, Z.I.; Kan, V.W.Y.; Ye, X.; Liebscher, S. Exciting Complexity: The Role of Motor Circuit Elements in ALS Pathophysiology. Front. Neurosci. 2020, 14, 573.
- Gunes, T.; Sirin, N.G.; Sahin, S.; Kose, E.; Isak, B. Use of CMAP, MScan fit-MUNE, and MUNIX in understanding neurodegeneration pattern of ALS and detection of early motor neuron loss in daily practice. Neurosci. Lett. 2020, 741, 135488.
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “Dying-Back” Phenomenon of Motor Neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477.
- Eisen, A.; Weber, M. The motor cortex and amyotrophic lateral sclerosis. Muscle Nerve 2001, 24, 564–573.
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic lateral sclerosis—A model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714.
- Baker, M.R. ALS—dying forward, backward or outward? Nat. Rev. Neurol. 2014, 10, 660.
- Grieve, S.M.; Menon, P.; Korgaonkar, M.S.; Gomes, L.; Foster, S.; Kiernan, M.C.; Vucic, S. Potential structural and functional biomarkers of upper motor neuron dysfunction in ALS. Amyotroph. Lateral Scler. Front. Degener. 2015, 17, 85–92.
- Geevasinga, N.; Van den Bos, M.; Menon, P.; Vucic, S. Utility of Transcranial Magnetic Simulation in Studying Upper Motor Neuron Dysfunction in Amyotrophic Lateral Sclerosis. Brain Sci. 2021, 11, 906.
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008, 131, 1540–1550.
- Kujirai, T.; Caramia, M.D.; Rothwell, J.C.; Day, B.L.; Thompson, P.D.; Ferbert, A.; Wroe, S.; Asselman, P.; Marsden, C.D. Corticocortical inhibition in human motor cortex. J. Physiol. 1993, 471, 501–519.
- Vucic, S.; Kiernan, M.C. Axonal excitability properties in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2006, 117, 1458–1466.
- Menke, R.A.L.; Agosta, F.; Grosskreutz, J.; Filippi, M.; Turner, M.R. Neuroimaging Endpoints in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2016, 14, 11–23.
- Vucic, S.; Pavey, N.; Haidar, M.; Turner, B.J.; Kiernan, M.C. Cortical hyperexcitability: Diagnostic and pathogenic biomarker of ALS. Neurosci. Lett. 2021, 759, 136039.
- Vucic, S.; van den Bos, M.; Menon, P.; Howells, J.; Dharmadasa, T.; Kiernan, M.C. Utility of threshold tracking transcranial magnetic stimulation in ALS. Clin. Neurophysiol. Pract. 2018, 3, 164–172.
- McMackin, R.; Dukic, S.; Costello, E.; Pinto-Grau, M.; McManus, L.; Broderick, M.; Chipika, R.; Iyer, P.M.; Heverin, M.; Bede, P.; et al. Cognitive network hyperactivation and motor cortex decline correlate with ALS prognosis. Neurobiol. Aging 2021, 104, 57–70.
- Hallett, M. Transcranial Magnetic Stimulation: A Primer. Neuron 2007, 55, 187–199.
- Triggs, W.J.; Menkes, D.; Onorato, J.; Yan, R.S.H.; Young, M.S.; Newell, K.; Sander, H.W.; Soto, O.; Chiappa, K.H.; Cros, D. Transcranial magnetic stimulation identifies upper motor neuron involvement in motor neuron disease. Neurology 1999, 53, 605.
- Agarwal, S.; Highton-Williamson, E.; Caga, J.; Howells, J.; Dharmadasa, T.; Matamala, J.M.; Ma, Y.; Shibuya, K.; Hodges, J.R.; Ahmed, R.M.; et al. Motor cortical excitability predicts cognitive phenotypes in amyotrophic lateral sclerosis. Sci. Rep. 2021, 11, 2172.
- Zanette, G.; Tamburin, S.; Manganotti, P.; Refatti, N.; Forgione, A.; Rizzuto, N. Changes in motor cortex inhibition over time in patients with amyotrophic lateral sclerosis. J. Neurol. 2002, 249, 1723–1728.
- Mills, K.R. Motor neuron disease. Brain 1995, 118, 971–982.
- Rösler, K.; Truffert, A.; Hess, C.; Magistris, M. Quantification of upper motor neuron loss in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2000, 111, 2208–2218.
- Prout, A.J.; Eisen, A.A. The cortical silent period and amyotrophic lateral sclerosis. Muscle Nerve 1994, 17, 217–223.
- Yokota, T.; Yoshino, A.; Inaba, A.; Saito, Y. Double cortical stimulation in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 1996, 61, 596–600.
- Ziemann, U.; Winter, M.; Reimers, C.D.; Reimers, K.; Tergau, F.; Paulus, W. Impaired motor cortex inhibition in patients with amyotrophic lateral sclerosis: Evidence from paired transcranial magnetic stimulation. Neurology 1997, 49, 1292–1298.
- Eisen, A.; Weber, M. Neurophysiological evaluation of cortical function in the early diagnosis of ALS. Amyotroph. Lateral Scler. 2000, 1, S47–S51.
- Turner, M.R.; Osei-Lah, A.D.; Hammers, A.; Al-Chalabi, A.; Shaw, C.; Andersen, P.M.; Brooks, D.; Leigh, P.N.; Mills, K.R. Abnormal cortical excitability in sporadic but not homozygous D90A SOD1 ALS. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1279–1285.
- Vucic, S.; Cheah, B.C.; Yiannikas, C.; Kiernan, M.C. Cortical excitability distinguishes ALS from mimic disorders. Clin. Neurophysiol. 2011, 122, 1860–1866.
- Menon, P.; Kiernan, M.C.; Vucic, S. Cortical hyperexcitability precedes lower motor neuron dysfunction in ALS. Clin. Neurophysiol. 2015, 126, 803–809.
- Tankisi, H.; Nielsen, C.S.-Z.; Howells, J.; Cengiz, B.; Samusyte, G.; Koltzenburg, M.; Blicher, J.U.; Møller, A.T.; Pugdahl, K.; Fuglsang-Frederiksen, A.; et al. Early diagnosis of amyotrophic lateral sclerosis by threshold tracking and conventional transcranial magnetic stimulation. Eur. J. Neurol. 2021, 28, 3030–3039.
- Werhahn, K.J.; Kunesch, E.; Noachtar, S.; Benecke, R.; Classen, J. Differential effects on motorcortical inhibition induced by blockade of GABA uptake in humans. J. Physiol. 1999, 517, 591–597.
- Ziemann, U.; Tergau, F.; Wassermann, E.M.; Wischer, S.; Hildebrandt, J.; Paulus, W. Demonstration of facilitatory I wave interaction in the human motor cortex by paired transcranial magnetic stimulation. J. Physiol. 1998, 511, 181–190.
- Ziemann, U.; Reis, J.; Schwenkreis, P.; Rosanova, M.; Strafella, A.; Badawy, R.; Müller-Dahlhaus, F. TMS and drugs revisited 2014. Clin. Neurophysiol. 2015, 126, 1847–1868.
- Menon, P.; Geevasinga, N.; Bos, M.V.D.; Yiannikas, C.; Kiernan, M.C.; Vucic, S. Cortical hyperexcitability and disease spread in amyotrophic lateral sclerosis. Eur. J. Neurol. 2017, 24, 816–824.
- Van den Bos, M.A.J.; Higashihara, M.; Geevasinga, N.; Menon, P.; Kiernan, M.C.; Vucic, S. Imbalance of cortical facilitatory and inhibitory circuits underlies hyperexcitability in ALS. Neurology 2018, 91, e1669–e1676.
- Pfurtscheller, G.; Lopes da Silva, F.H. Event-related EEG/MEG synchronization and desynchronization: Basic principles. Clin. Neurophysiol. 1999, 110, 1842–1857.
- Kasahara, T.; Terasaki, K.; Ogawa, Y.; Ushiba, J.; Aramaki, H.; Masakado, Y. The correlation between motor impairments and event-related desynchronization during motor imagery in ALS patients. BMC Neurosci. 2012, 13, 66.
- Bizovičar, N.; Dreo, J.; Koritnik, B.; Zidar, J. Decreased movement-related beta desynchronization and impaired post-movement beta rebound in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2014, 125, 1689–1699.
- Riva, N.; Falini, A.; Inuggi, A.; Gonzalez-Rosa, J.; Amadio, S.; Cerri, F.; Fazio, R.; Del Carro, U.; Comola, M.; Comi, G.; et al. Cortical activation to voluntary movement in amyotrophic lateral sclerosis is related to corticospinal damage: Electrophysiological evidence. Clin. Neurophysiol. 2012, 123, 1586–1592.
- Proudfoot, M.; Rohenkohl, G.; Quinn, A.; Colclough, G.L.; Wuu, J.; Talbot, K.; Woolrich, M.W.; Benatar, M.; Nobre, A.C.; Turner, M.R. Altered cortical beta-band oscillations reflect motor system degeneration in amyotrophic lateral sclerosis. Hum. Brain Mapp. 2016, 38, 237–254.
- Logothetis, N.K. What we can do and what we cannot do with fMRI. Nature 2008, 453, 869–878.
- Hermes, D.; Nguyen, M.; Winawer, J. Neuronal synchrony and the relation between the blood-oxygen-level dependent response and the local field potential. PLoS Biol. 2017, 15, e2001461.
- Konrad, C.; Jansen, A.; Henningsen, H.; Sommer, J.; Turski, P.A.; Brooks, B.R.; Knecht, S. Subcortical reorganization in amyotrophic lateral sclerosis. Exp. Brain Res. 2006, 172, 361–369.
- Schoenfeld, M.A.; Tempelmann, C.; Gaul, C.; Kühnel, G.R.; Düzel, E.; Hopf, J.-M.; Feistner, H.; Zierz, S.; Heinze, H.-J.; Vielhaber, S. Functional motor compensation in amyotrophic lateral sclerosis. J. Neurol. 2005, 252, 944–952.
- Douaud, G.; Filippini, N.; Knight, S.; Talbot, K.; Turner, M. Integration of structural and functional magnetic resonance imaging in amyotrophic lateral sclerosis. Brain 2011, 134, 3470–3479.
- Agosta, F.; Spinelli, E.G.; Marjanovic, I.V.; Stevic, Z.; Pagani, E.; Valsasina, P.; Salak-Djokic, B.; Jankovic, M.; Lavrnic, D.; Kostic, V.S.; et al. Unraveling ALS due to SOD1 mutation through the combination of brain and cervical cord MRI. Neurology 2018, 90, e707–e716.
- Li, W.; Zhang, J.; Zhou, C.; Hou, W.; Hu, J.; Feng, H.; Zheng, X. Abnormal Functional Connectivity Density in Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2018, 10, 215.
- Commisso, B.; Ding, L.; Varadi, K.; Gorges, M.; Bayer, D.; Boeckers, T.M.; Ludolph, A.C.; Kassubek, J.; Müller, O.J.; Roselli, F. Stage-dependent remodeling of projections to motor cortex in ALS mouse model revealed by a new variant retrograde-AAV9. eLife 2018, 7, e36892.
- Proudfoot, M.; Colclough, G.L.; Quinn, A.; Wuu, J.; Talbot, K.; Benatar, M.; Nobre, A.C.; Woolrich, M.W.; Turner, M.R. Increased cerebral functional connectivity in ALS. Neurology 2018, 90, e1418–e1424.
- Foerster, B.R.; Callaghan, B.C.; Petrou, M.; Den, R.A.E.; Chenevert, T.L.; Feldman, E.L. Decreased motor cortex γ-aminobutyric acid in amyotrophic lateral sclerosis. Neurology 2012, 78, 1596–1600.
- Foerster, B.R.; Pomper, M.G.; Callaghan, B.C.; Petrou, M.; Edden, R.; Mohamed, M.; Welsh, R.C.; Carlos, R.C.; Barker, P.B.; Feldman, E. An Imbalance Between Excitatory and Inhibitory Neurotransmitters in Amyotrophic Lateral Sclerosis Revealed by Use of 3-T Proton Magnetic Resonance Spectroscopy. JAMA Neurol. 2013, 70, 1009–1016.
- Petri, S.; Krampfl, K.; Hashemi, F.; Grothe, C.; Hori, A.; Dengler, R.; Bufler, J. Distribution of GABAAReceptor mRNA in the Motor Cortex of ALS Patients. J. Neuropathol. Exp. Neurol. 2003, 62, 1041–1051.
- Aronica, E.; Baas, F.; Iyer, A.; Asbroek, A.L.T.; Morello, G.; Cavallaro, S. Molecular classification of amyotrophic lateral sclerosis by unsupervised clustering of gene expression in motor cortex. Neurobiol. Dis. 2014, 74, 359–376.
- Shepherd, G.M.G. Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci. 2013, 14, 278–291.
- Nigro, M.J.; Hashikawa-Yamasaki, Y.; Rudy, B. Diversity and Connectivity of Layer 5 Somatostatin-Expressing Interneurons in the Mouse Barrel Cortex. J. Neurosci. 2018, 38, 1622–1633.
- Swanson, O.K.; Maffei, A. From Hiring to Firing: Activation of Inhibitory Neurons and Their Recruitment in Behavior. Front. Mol. Neurosci. 2019, 12, 168.
- Rudy, B.; Fishell, G.; Lee, S.; Hjerling-Leffler, J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 2011, 71, 45–61.
- Tremblay, R.; Lee, S.; Rudy, B. GABAergic interneurons in the neocortex: From cellular properties to circuits. Neuron 2016, 91, 260–292.
- Wood, K.; Blackwell, J.M.; Geffen, M.N. Cortical inhibitory interneurons control sensory processing. Curr. Opin. Neurobiol. 2017, 46, 200–207.
- Lee, S.; Kruglikov, I.; Huang, Z.J.; Fishell, G.; Rudy, B. A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat. Neurosci. 2013, 16, 1662–1670.
- Yu, J.; Hu, H.; Agmon, A.; Svoboda, K. Recruitment of GABAergic interneurons in the barrel cortex during active tactile behavior. Neuron 2019, 104, 412–427.e414.
- Gu, Q. Neuromodulatory transmitter systems in the cortex and their role in cortical plasticity. Neuroscience 2002, 111, 815–835.
- Conner, J.M.; Kulczycki, M.; Tuszynski, M.H. Unique Contributions of Distinct Cholinergic Projections to Motor Cortical Plasticity and Learning. Cereb. Cortex 2010, 20, 2739–2748.
- Vitrac, C.; Benoit-Marand, M. Monoaminergic Modulation of Motor Cortex Function. Front. Neural Circuits 2017, 11, 72.
- Fogarty, M.J.; Mu, E.W.H.; Noakes, P.; Lavidis, N.A.; Bellingham, M.C. Marked changes in dendritic structure and spine density precede significant neuronal death in vulnerable cortical pyramidal neuron populations in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2016, 4, 77.
- Fogarty, M.J.; Noakes, P.; Bellingham, M.C. Motor Cortex Layer V Pyramidal Neurons Exhibit Dendritic Regression, Spine Loss, and Increased Synaptic Excitation in the Presymptomatic hSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurosci. 2015, 35, 643–647.
- Fogarty, M.J.; Klenowski, P.M.; Lee, J.D.; Drieberg-Thompson, J.R.; Bartlett, S.E.; Ngo, S.T.; Hilliard, M.A.; Bellingham, M.C.; Noakes, P.G. Cortical synaptic and dendritic spine abnormalities in a presymptomatic TDP-43 model of amyotrophic lateral sclerosis. Sci. Rep. 2016, 6, 37968.
- Kim, J.; Hughes, E.G.; Shetty, A.S.; Arlotta, P.; Goff, L.; Bergles, D.E.; Brown, S.P. Changes in the Excitability of Neocortical Neurons in a Mouse Model of Amyotrophic Lateral Sclerosis Are Not Specific to Corticospinal Neurons and Are Modulated by Advancing Disease. J. Neurosci. 2017, 37, 9037–9053.
- Buskila, Y.; Kékesi, O.; Bellot-Saez, A.; Seah, W.; Berg, T.; Trpceski, M.; Yerbury, J.; Ooi, L. Dynamic interplay between H-current and M-current controls motoneuron hyperexcitability in amyotrophic lateral sclerosis. Cell Death Dis. 2019, 10, 310.
- Saba, L.; Viscomi, M.T.; Martini, A.; Caioli, S.; Mercuri, N.B.; Guatteo, E.; Zona, C. Modified age-dependent expression of NaV1.6 in an ALS model correlates with motor cortex excitability alterations. Neurobiol. Dis. 2019, 130, 104532.
- Dyer, M.S.; Reale, L.A.; Lewis, K.E.; Walker, A.K.; Dickson, T.C.; Woodhouse, A.; Blizzard, C.A. Mislocalisation of TDP-43 to the cytoplasm causes cortical hyperexcitability and reduced excitatory neurotransmission in the motor cortex. J. Neurochem. 2020, 157, 1300–1315.
- Schmitt-John, T.; Drepper, C.; Mußmann, A.; Hahn, P.; Kuhlmann, M.; Thiel, C.; Hafner, M.; Lengeling, A.; Heimann, P.; Jones, J.M.; et al. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat. Genet. 2005, 37, 1213–1215.
- Nieto-Gonzalez, J.L.; Moser, J.; Lauritzen, M.; Schmitt-John, T.; Jensen, K. Reduced GABAergic Inhibition Explains Cortical Hyperexcitability in the Wobbler Mouse Model of ALS. Cereb. Cortex 2011, 21, 625–635.
- Scekic-Zahirovic, J.; Sanjuan-Ruiz, I.; Kan, V.; Megat, S.; De Rossi, P.; Dieterlé, S.; Cassel, R.; Jamet, M.; Kessler, P.; Wiesner, D.; et al. Cytoplasmic FUS triggers early behavioral alterations linked to cortical neuronal hyperactivity and inhibitory synaptic defects. Nat. Commun. 2021, 12, 3028.
- Khademullah, C.S.; Aqrabawi, A.J.; Place, K.M.; Dargaei, Z.; Liang, X.; Pressey, J.C.; Bedard, S.; Yang, J.W.; Garand, D.; Keramidis, I.; et al. Cortical interneuron-mediated inhibition delays the onset of amyotrophic lateral sclerosis. Brain 2020, 143, 800–810.
- Özdinler, P.H.; Benn, S.; Yamamoto, T.H.; Güzel, M.; Brown, R.H.; Macklis, J.D. Corticospinal Motor Neurons and Related Subcerebral Projection Neurons Undergo Early and Specific Neurodegeneration in hSOD1G93A Transgenic ALS Mice. J. Neurosci. 2011, 31, 4166–4177.
- Clark, R.M.; Blizzard, C.A.; Young, K.M.; King, A.E.; Dickson, T.C. Calretinin and Neuropeptide Y interneurons are differentially altered in the motor cortex of the SOD1G93A mouse model of ALS. Sci. Rep. 2017, 7, 44461.
- Brunet, A.; Stuart-Lopez, G.; Burg, T.; Scekic-Zahirovic, J.; Rouaux, C. Cortical Circuit Dysfunction as a Potential Driver of Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 363.
- Takahashi, H.; Snow, B.J.; Bhatt, M.H.; Peppard, R.; Eisen, A.; Calne, D.B. Evidence for a dopaminergic deficit in sporadic amyotrophic lateral sclerosis on positron emission scanning. Lancet 1993, 342, 1016–1018.
- Borasio, G.D.; Linke, R.; Schwarz, J.; Schlamp, V.; Abel, A.; Mozley, P.D.; Tatsch, K. Dopaminergic deficit in amyotrophic lateral sclerosis assessed with IPT single photon emission computed tomography. J. Neurol. Neurosurg. Psychiatry 1998, 65, 263–265.
- Kostic, V.; Gurney, M.E.; Deng, H.-X.; Siddique, T.; Epstein, C.J.; Przedborski, S. Midbrain dopaminergic neuronal degeneration in a transgenic mouse model of familial amyotrophic lateral sclerosis. Ann. Neurol. 1997, 41, 497–504.
- Dentel, C.; Palamiuc, L.; Henriques, A.; Lannes, B.; Spreux-Varoquaux, O.; Gutknecht, L.; René, F.; Echaniz-Laguna, A.; De Aguilar, J.-L.G.; Lesch, K.P.; et al. Degeneration of serotonergic neurons in amyotrophic lateral sclerosis: A link to spasticity. Brain 2013, 136, 483–493.
This entry is offline, you can click here to edit this entry!