Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Health Care Sciences & Services
Alzheimer’s disease (AD) is the most common neurodegenerative dementia, affecting two-thirds of individuals with cognitive decline worldwide. Several links between vascular risk factors (VRF), neurovascular unit dysfunction (NVUd), blood-brain barrier breakdown (BBBb) and AD onset and progression in adulthood, suggesting a pathogenetic continuum between AD and vascular dementia. Shared pathways between AD, VRF, and NVUd/BBBb have also been found at the molecular level, underlining the strength of this association.
- Alzheimer’s disease
- vascular risk factors
- hypertension
- smoking
- dyslipidemia
- diabetes mellitus
- neurovascular unit
- blood brain barrier breakdown
1. Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative dementia, affecting two-thirds of individuals with cognitive decline worldwide [1]. Its main pathological features are represented by neuroinflammation, extracellular amyloid-β (Aβ) peptide deposition, intracellular neurofibrillary tangles, tau protein degeneration, and neural loss with progressive deterioration of cognitive function [2,3,4]. Considering a doubling in 20 years, the prevalence of AD will reach 130 million people in 2050, with the greatest increase expected in the poorest countries [5]. Due to its high prevalence, several studies are focusing on reliable serum biomarkers to accurately diagnose AD [3,4].
The neuropathology of AD is characterised by structural and physiological changes that may involve different brain areas. This variability contributes to a certain heterogeneity in the final clinical manifestations in AD patients, each of whom may exhibit a variable association of different neuropsychological deficits. In fact, while the classic cognitive profile of AD is mainly characterised by episodic memory deficits due to the impairment of the temporal lobe, several recent studies have shown that in relation to the different brain areas predominantly involved, different clinical pictures may be present. For example, alterations in the medial prefrontal cortex are associated with impaired retrieval and extinction memories, whereas impairment of emotional and executive processing, similarly to the psychiatric population, reflects a probable impairment of the lateral orbitofrontal cortex or the inferior frontal gyrus [6,7,8]. It is worth underlining that multiple pathways are implicated in the onset and progression of neurodegeneration and specific functions, such as emotional control, are often impaired in dementia. Indeed, impairment of the prefrontal cortex causes in AD patients an impairment in emotion processing that impacts on action and motor control [7].
The AD pathophysiology is considered largely heterogeneous and characterized by both neurodegeneration—characterized by aberrant, misfolded and aggregated Aβ [9] and hyperphosphorylated tau proteins [10]—and vascular disease, with a common involvement of large [11,12,13,14] and small brain vessels. Other neurotoxic elements could also play a relevant role: oxidative stress with reactive oxygen species overproduction, mitochondrial dysfunction or metal accumulation have been extensively studied in the last years [15].
Recently a great attention was put on the interaction between neuronal (neurons and glia) and vascular tissues (endothelial cells, pericytes, and adventitial cells) that are functionally organized in the neurovascular unit (NVU). The NVU is responsible for so-called neurovascular coupling, an organized vascular response to specific neuronal stimuli aimed at modifying regional cerebral blood flow and neuronal metabolic activity [16]. The blood-brain barrier (BBB) is a part of the NVU that controls the transfer of molecules and pathogens to and from brain tissue by adopting a specific transport system in brain endothelial cells [17]. The BBB transports brain metabolic waste products from the brain interstitial fluid to the bloodstream. Thus, BBB represents the most important site of metabolic homeostasis in the central nervous system [17]. Neurovascular coupling is typically deranged in several pathological conditions such as hypertension [18], acute ischemic stroke [19] and AD [20], suggesting a potential role of NVU dysfunction (NVUd) in the progression of cognitive impairment. Of note, neurovascular coupling is strongly influenced by VRF [21,22,23,24,25,26] and atherosclerotic vascular pathology [14,27], as synthesized in Figure 1.
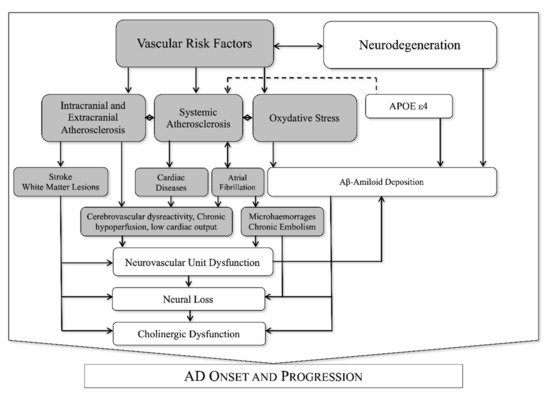
Figure 1. Known interactions between vascular and neurodegenerative factors in AD.
Postmortem studies emphasize an important role of vascular pathology in a large percentage of AD subjects. As first observed in the Nun study [6], the presence of neuropathologic findings of vascular lesions in AD subjects was associated with a history of worse cognitive performances. In addition, other studies have confirmed the presence of atherosclerosis of large and small vessels in AD [12]. These alterations, which could affect NVU activities, may be expressed by pathologic adaptations of cerebrovascular responsiveness to hypercapnia [26,28]. A dysfunction of the BBB has been associated with oxidative stress [29], advanced glycation end products (AGEs) and their receptor (RAGE) [30,31] and increased production of proinflammatory cytokines [32]. A blood-brain barrier breakdown (BBBb) could also contribute to AD onset and progression.
2. Type 2 Diabetes Mellitus
2.1. The Clinical and Epidemiological Link between AD and T2DM
Both AD and T2DM prevalence are progressively increasing, especially among elderly patients [1,33]. Among adults, 1 in 11 suffers from diabetes mellitus and 90% of cases are T2DM, which is a chronic, multi-organ disease characterised by a high burden of co-morbidities and a low quality of life [34]. While aging itself is the strongest risk factor for both AD [35] and T2DM [36], emerging epidemiological data suggest that T2DM and other VRFs may contribute to the pathogenesis of AD directly, in association, or as cofactors [37,38,39]. AD and T2DM are epidemiologically associated as AD patients appear more vulnerable to T2DM [40], and individuals with T2DM show an increased risk of dementia, including AD [41].
The Rotterdam Study confirmed that the presence of T2DM increases the risk of AD [42], and that this association is stronger in patients with a long history of T2DM. This epidemiological relationship has been confirmed in other cohorts [43]. However, despite epidemiological studies suggesting T2DM as a potential risk factor for AD, the demonstration of a complete overlap between the two diseases is lacking. Some authors have associated this effect to different insulin resistance in target organs. Cerebral hyperglycemia [44] in the absence of clinically evident T2DM has been positively associated with accelerated cognitive impairment, even adjusting for other risk factors, including age and macrovascular disease. In addition, patients with AD often exhibit both insulin resistance and insulin insufficiency even when not affected by T2DM. These observations have led to the current concept of AD as a special form of T2DM, defined by several authors as “type 3 diabetes mellitus” (T3DM) [45,46,47]. T3DM refers to insulin/insulin-like growth factor (IGF) deficiency and insulin/IGF resistance in brain tissue [46].
2.2. The Role of Insulin Signalling
Insulin/IGF and their receptors are widely expressed in the cortex, hippocampus, and hypothalamus of the human brain. Several investigations support the hypothesis that cognitive impairment in AD might be, at least in part, mediated by insulin resistance and deficiency of the insulin/IGF cascade in the brain [48,49,50]. These mechanisms activate multiple intracellular signalling pathways ensuing in intrinsic tyrosine kinases (iTK) activation starting with ligand binding to cell surface receptors, followed by iTK autophosphorylation and activation [51,52]. iTK phosphorylate IRS molecules [53,54,55], which transmit signals downstream by activating the extracellular/mitogen-activated signal-related kinase (ERK/MAPK) and PI3K pathways and inhibit glycogen synthase kinase 3beta (GSK3). PI3K/AKT/mTOR cascade activation leads to synaptic formation, increased neuronal cell survival [56], regional vasodilation, and regulation of cerebrovascular reactivity in the neurovascular unit [57].
Postmortem studies pointed out that, in brain samples from AD patients, insulin and insulin receptor expression were severely impaired and their levels were inversely proportional to the extent of neurodegeneration [45,58] along with impairment of insulin receptor binding capacity and reduced expression of insulin, IGF-1, IGF2 mRNA and their receptors, with a reduction in the cytosolic level of PI3K p85a and p110a subunits [59]. This was consensual with a tau protein reduction, regulated by insulin/IGF-1.
Decreased choline acetyltransferase (ChAT) expression, typically described in AD, is associated with reduced ChAT colocalization with the insulin/IGF-1 receptor, confirming that neuronal expression of tau and ChAT is regulated by insulin/IGF-1 in the human brain [46,47]. Reduced insulin and poor insulin receptor sensitivity contribute to decreased acetylcholine (ACh), further elucidating a possible biochemical link between diabetes and AD [58]. Thus, insulin resistance and deficiency in the brain could explain, at least in part, the alterations observed in AD, such as cytoskeletal collapse, retraction of neurites, synaptic disconnection, loss of neuronal plasticity, and deficiencies in ACh production. Moreover, T2DM is known to be one of the most important factors for accelerated atherosclerosis [60], and these observations suggest that cerebral vessel atherosclerosis could be another potential link between the two diseases, as confirmed by clinical studies [14].
2.3. Shared Molecular Mechanisms between AD and T2DM
Primary biological responses to insulin/IGF include increase in cell growth, survival, energy metabolism and cholinergic gene expression, and inhibition of oxidative stress and of apoptosis. These signalling pathways are activated in different cell types and tissues capable of expressing insulin/IGF receptor. Thus they are virtually universal [55,61,62,63]. Several authors enlightened different abnormalities in IRS-1 phosphorylation (IRS-1p) in AD brains [64]. IRS-1p on tyrosine residues is needed for insulin-stimulated responses, whereas IRS-1p on serine residues was associated to an insulin reduced response, which was consistent with insulin resistance [65].
This pathway modulates the expression of Aβ precursor protein (APP), kinesin, Abelson helper integration site-1 (AHI-1), huntingtin-associated protein-1 (HAP-1), and tau, which are all involved in the neuropathology of AD. Furthermore, neuronal and oligodendroglial cell survival and function are fully linked to the integrity of the insulin/IGF-1 pathway [46,47,49]. Impairment of these metabolic pathways leads to deficits in energy metabolism resulting in increased oxidative stress, mitochondrial dysfunction, activation of proinflammatory cytokines and APP expression. Consequently, reduced expression of neuronal and oligodendroglial specific genes and increased expression of astrocytic and microglial inflammatory genes in AD have been attributed to progressive brain insulin/IGF deficiency and resistance.
Microglial and astrocytic APP mRNA levels are increased in the early stages of neurodegeneration in AD [66]. Microglia activation promotes APP gene expression, cleavage and accumulation. Impairment of insulin/IGF signalling leads to oxidative stress and mitochondrial dysfunction that induces APP gene expression and cleavage, thus resulting in neurotoxicity due to APP-A accumulation. Tau gene expression and tau protein phosphorylation are specifically mediated by this signalling cascade [67,68].
2.4. The Role of AGE/RAGE System in AD
Glycosylation is a non-reversible and non-enzymatic reaction that occurs between proteins and glucose and eventually leads to the production of AGEs [69], which is especially observed in subjects affected by complicated T2DM [70]. The presence of AGEs marginally affects cell survival, but can significantly alter neuronal metabolism and thus brain function in several neurodegenerative disorders, such as AD [71,72]. In addition, AGEs can directly induce oxidative stress and promote the release of proinflammatory cytokines, thereby worsening cognitive dysfunction in AD [73]. AGEs and RAGEs colocalize with Aβ, senile plaques, and neurofibrillary tangles. Specifically, the interaction between RAGE and Aβ activates neuroinflammatory signalling pathways, causes the release of reactive oxygen species, and ultimately induces neuronal and mitochondrial dysfunction [31].
However, one large study observed a lack of longitudinal association between AGE-RAGE system dysfunction and dementia, suggesting a potential short-term association or reverse causality [74], thus supporting the need for further studies to explore this association.
3. Hypertension
3.1. The Clinical and Epidemiological Link between AD and Hypertension
Aging is an important risk factor for hypertension, representing one of the epidemiological links between AD and VRF. However, the clinical association between hypertension and AD seems weaker than that with T2DM. Some studies have described this potential association [75,76,77] while others failed to demonstrate any link [78,79]. Papers underlining this epidemiological link have longer follow-up times [75,76,77]. The Rotterdam study pointed out that hypertension preceded the onset of AD by nine years [75], while in the Honolulu-Asia Aging Study, a temporal relationship of 20–26 years was observed [77].
On the other hand, some studies have shown that low blood pressure is also associated with incident dementia, and that blood pressure drops in the preclinical stages of AD, during AD, and consistently with advanced cognitive impairment [80]. Recently, a U-shaped relationship between hypertension and cognition has been confirmed, especially among the elderly [81]. Several authors have suggested that this effect might be mediated by neurodegeneration of brain structures involved in the central regulation of blood pressure (hypothalamus, amygdala, insular cortex, medial prefrontal cortex, locus coeruleus, parabrachial nucleus, pons and medulla oblongata). This hypothesis is supported by studies underlining a direct positive relationship between neuron number in pons or medulla and blood pressure in AD [82]. Further, brain atrophy has been correlated with lower blood pressure in elderly patients, regardless of dementia [83]. In addition, lower blood pressure can result in greater neuronal damage by worsening regional cerebral blood flow regulation by generating local hypoperfusion [84].
Thus, middle-aged hypertension acts as a risk factor for AD before its onset, whereas low blood pressure in the elderly should be interpreted as a consequence of neural loss, especially in advanced AD. Hypertension is known to induce cerebral vascular changes and vascular dementia. Animal models have shown that high blood pressure can also lead to AD-like neuropathology [85,86,87], with accumulation and deposition of Aβ.
3.2. Shared Molecular Mechanisms between AD and Hypertension
While the “classical” amyloid hypothesis suggested a cytotoxic accumulation of Aβ in the brain tissue of AD patients due to its overproduction [88], more recent evidence has shown that Aβ accumulation might be more related to an altered clearance of this molecule from the BBB due to NVU dysfunction [89].
One of the pathways linking hypertension to AD is RAGE, which modulates Aβ clearance in the BBB. Its expression is critically increased in endothelial cells and at the level of the AD brain neurovascular unit [90]. Furthermore, in experimental models, its expression is upregulated in cerebral vessels of the cortex and hippocampus after exposure to a hypertensive condition [86]. RAGE acts as a scavenger receptor for Aβ. In the BBB it mediates the passage of Aβ from the blood to the brain. It also stimulates Aβ production [91] and induces tau hyperphosphorylation [92] by activating the GSK-3 cascade. The angiotensin-II type 1 receptor suggests another link between AD and hypertension. In hypertensive subjects, activation of this receptor increased RAGE mRNA expression, suggesting a link between activation of the renin-angiotensin axis and AD progression [93]. RAGE also responds to AGEs, which are elevated in AD, especially in patients with T2DM, and this represents another possible link between VRF and AD.
The other molecular mechanism linking AD to hypertension is low-density lipoprotein receptor-related protein-1 (LRP-1). Most cell types in the neurovascular unit express LRP-1, which is able to maintain the BBB integrity and transport Aβ from the brain to the blood vessels, in a direction opposite to that by RAGE. LRP-1 acts primarily by releasing Aβ from the brain, but its soluble form (sLRP-1) can bind Aβ and remove it from the circulation, reducing its bioavailability. Interestingly, the expression of LRP-1 in endothelial cells of the neurovascular unit is reduced with aging and its activity is mediated by ApoE [94]. In murine models of hypertension, RAGE expression is increased, whereas LRP-1 expression is unchanged, suggesting increased Aβ influx that is not adequately counteracted by increased efflux [95]. Furthermore, the presence of oxidative stress, as commonly observed in association with the presence of VRF, decreases sLRP-1 activity and increases serum levels of Aβ, which negatively correlates with cognition [96].
Finally, hypertensive patients often show increased serum levels of several markers of endothelial damage, such as soluble intercellular adhesion molecule-1 (sICAM-1), soluble vascular cell adhesion molecule-1 (sVCAM-1), and endothelin-1 (ET-1), which might be implicated in the dysregulation of cerebrovascular reactivity in AD and other neurodegenerative diseases by promoting vasoconstriction [97,98,99].
4. Dyslipidaemia
4.1. The Clinical and Epidemiological Link between AD and Dyslipidaemia
Dyslipidaemias are a heterogeneous group of diseases defined as disorders of lipid metabolism that lead, alone or in association with other VRFs, to cerebral and systemic atherosclerosis. The current management of dyslipidaemias is closely dependent on the presence and extent of other VRFs, and current guidelines suggest treatment according to the patient’s overall cardiovascular risk, as assessed by formal scores [100]. The systematic assessment and proper management of cardiovascular risk is leading to a progressive reduction in the incidence of atherosclerosis. However, this disease remains one of the leading causes of mortality and morbidity worldwide [100]. The link between dyslipidaemia and AD has been described at several levels. Epidemiological evidence suggests an association between high serum cholesterol levels and AD, with a potential role for lipids in modulating AD expression. Total cholesterol serum levels appear to be independently associated with increased AD prevalence, with a potential modulation of the effect by ApoE genotype [101]. Similar to hypertension, increased serum total cholesterol in middle age also appears to be strongly associated with the risk of AD, with a 3-fold increase in the likelihood of development, independent of ApoE genotype [102]. High levels of LDL cholesterol (LDL-C) correlate with lower global cognition in the absence of clinical dementia [103], and with more rapid cognitive decline in individuals who will develop AD [104]. Some authors underlined a paradoxically protective effect of increased serum cholesterol levels from dementia in late life [105], underlining the detrimental role of dyslipidaemia in younger subjects [106]. In addition, intracranial and extracranial atherosclerosis, one of the major consequences of inadequately treated dyslipidaemia, is significantly associated with the risk of AD onset and progression [14,107].
4.2. Shared Molecular Mechanisms between AD and Dyslipidaemia
Increased serum cholesterol levels are presumed to induce neuronal apoptosis, oxidative stress, and tau hyperphosphorylation [108]. Brain lipid composition appears to be directly involved in APP processing and Aβ production: increased endosomal cholesterol levels appear to unbalance APP processing, thereby promoting the amyloid-genic pathway [109,110]. A cholesterol-rich membrane might also alter the activity of membrane secretases, thus inducing Aβ production [111]. Furthermore, dyslipidemia is thought to be associated with BBB disruption, which is commonly observed in AD [112]. Animal models, particularly low-density lipoprotein receptor (LDL-R) knock-out mice, confirm these observations: dyslipidemia increases the severity of cognitive dysfunction, especially learning and memory, and Aβ-associated neurotoxicity [113].
Recent studies have emphasized a genetic overlap between AD, C-reactive protein, and plasma lipids [114]. Genome-wide association studies emphasize a strong association between dyslipidemia and AD in several genes. ApoE genotype has been confirmed central to this interaction [115]. ApoE is the most abundant apolipoprotein in the human brain whose role is to transport lipids and facilitate brain homeostasis by removing debris from the interstitial fluid of the brain by interacting with endothelial cells, the basement membrane and glia [116]. In AD, APOE promotes Aβ clearance. The efficiency of Aβ clearance through the BBB depends on the activity of transport proteins such as APOE and APOJ, and receptors such as LRP-1 and RAGE. In particular, it has been observed that APOE ε2 and APOE ε3 genotypes bind with high affinity with LRP-1, whereas APOE ε4 binds with LDL-R [117]. The lack of interaction between APOE ε4 and LRP-1 has been associated with reduced cyclophilin A (CypA) inhibition leading to a proinflammatory state and BBB breakdown [118]. This effect appears to be mediated in pericytes by an NFB-dependent matrix metalloproteinase 9 (MMP-9), which disrupts endothelial tight junctions [117,119]. In addition, CypA has been associated with systemic atherosclerosis [117].
Other single nucleotide polymorphisms of genes implicated in lipid metabolism, such as CLU and ABCA7, have been associated with AD, underscoring a strong link between lipid homeostasis and cognitive function [120]. Of note, several genes implicated in the modulation of inflammation, such as CR1, HLA-DRB5 and TREM, have also been identified as associated with AD [120].
5. Cigarette Smoking
5.1. The Clinical and Epidemiological Link between AD and Cigarette Smoking
Some cross-sectional studies, supported by the tobacco industry, reported a lower AD prevalence among smokers [121]. However, when analysing incident cases and controlling for tobacco industry affiliation [122], it was observed that smoking consistently increased the risk for AD and cognitive decline [123]. This increased risk was found in both APOE ε4 allele carriers [124] and non-carriers [125]. Particularly, mid-life smoking was associated to an increased AD risk [126]. Smoking habit shows its detrimental effects in cognition at different levels. Compared to non-smokers, middle-aged, active smokers showed poorer neurocognitive performances in executive domains (processing speed, learning and memory). Such cognitive dysfunctions were associated with a reduced volume and thickness in hippocampal, cortical, and subcortical areas, reduced neuronal and BBB integrity and neurobiological alterations like those found in early-stage AD, with a dose-dependent effect. Elderly, active-smoking subjects showed worse executive functions, processing speed, learning and memory, a greater cortical atrophy and lower grey matter density in specific brain areas when compared to non-smokers. Former smokers showed intermediate abnormalities between smokers and non-smokers. Patients with chronic obstructive pulmonary disease (COPD), which is commonly caused by smoking, often show worse cognitive performances [127] that seem to be partially preserved by long-term oxygen therapy [128]. A midlife COPD diagnosis is associated to an increased risk of a later-life cognitive deterioration [129]. However, COPD and lung function impairment seem to affect only marginally incident AD [130].
5.2. Shared Molecular Mechanisms between AD and Cigarette Smoking
The only potential neuroprotective effect of smoking on the brain relies on the finding that nicotine showed neuroprotective activity against glutamate toxicity via α4 and α7 subunits, which can inhibit the neuronal apoptosis process similarly to therapeutic acetylcholinesterase inhibitors [131]. Cigarette smoking, however, has been associated to a downregulation of nicotinic acetylcholine receptors (nAChrs) subunit α7 expression on astrocytes [132] with a reduction of the neuroprotection offered. Furthermore, nicotine strongly affects brain endothelial function, since brain endothelium expresses several nicotinic receptor subunits (α3, α5, α7, β2 and β3) [133]. Nicotine increases BBB permeability by reducing tight junctions expression [134]. The detrimental effects of nicotine on tight junctions’ permeability are worsened by oxidative stress and hypoxia. Moreover, nicotine downregulates NOTCH-4 expression in brain endothelial cells: a reduced NOTCH-4 expression is also associated to BBB breakdown [133]. Chronic cigarette smoking has been associated to an increased Aβ deposition and amyloid burden, tau phosphorylation, neuroinflammation with microglial activation, and plaque formation in a dose-dependent manner [135]. On the other side, it has been demonstrated that different central nervous system cells express nicotinic subunits (α3, α4, α5, α6, α7, β2, β4) in the context of nAChrs with a wide variability of expression within different areas of the brain.
Smoking attitude increases oxidative stress by unbalancing the production of reactive oxygen/nitrogen species and their reduction by natural antioxidants [136,137]. Notably, oxidative damage acts on nucleic acids, proteins and lipid membranes of NVU cells [136,137]. Oxidative stress induces cytokine-mediated activation of inflammation in the NVU, thus inducing neuronal cell death and BBBb.
There is a narrow link between neurodegenerative diseases, as AD, and chronic lung pathologies, as COPD [138]. Chronic brain hypoxia, which is commonly observed in advanced COPD and worsened by the occurrence of significant carotid atherosclerosis, seems to worsen cognition by increasing Aβ deposition and tau hyperphosphorylation [139,140]. Moreover, chronic hypoxia acts on VMSCs by downregulating LRP-1 [140], favouring a hypercontractile, non Aβ-clearing phenotype [141]. Moreover, chronic hypoxia has other detrimental effects on cognition: it activates microglia inducing a proinflammatory state that downregulates Aβ receptors and induces a BBB breakdown [140].
6. Association between VRF and NVU Dysfunction in AD
Neurovascular imbalance is sufficient to initiate neuronal damage and induce accumulation of Aβ. VRF aggregation leads to atherosclerosis, and both factors can induce NVU dysfunction and BBB disruption. These two alterations are associated with increased entry and defective clearance of neurotoxic compounds into brain tissues and reduced energy metabolites and oxygen delivery to activated areas of the brain resulting in neuronal damage. Furthermore, by regulating small vessel blood flow, neurovascular coupling aims to reduce local thrombosis by balancing pro-thrombotic and anti-thrombotic pathways. Its alteration is associated with increased vascular damage. Different combinations of VRF have been associated with cognitive impairment [26,39], especially in the presence of altered cerebrovascular reactivity [142]. Large vessels atherosclerosis is the most prominent effect of a long-term VRF combination, has been associated with an imbalance in cerebrovascular reactivity and more rapid cognitive deterioration [27,143].
At the cellular level, vascular muscle smooth cells (VMSCs) in AD exhibit a “hypercontractile phenotype,” which appears to be critically involved in the dysregulation of local cerebral blood flow by inducing chronic hypoxia and hypoperfusion that facilitates neural loss [141]. In addition, AD-VMSCs exhibit impaired capacity to clear Aβ, facilitating cerebral amyloid angiopathy, which in turn leads to impaired cerebral hemodynamic adaptability [144].
BBB disruption appears to be associated with increased production and reduced clearance of Aβ, which promotes the accumulation of amyloidogenic molecules, typically present in advanced AD. BBB dysfunction is favoured by genetic traits, such as APOE ε4. APOE ε4 carriers show accelerated pericyte degeneration due to activation of the CypA MMP-9 pathway, which is associated with BBB dysfunction with tight junctions and alteration of core proteins [145]. In addition, APOE ε4 carriers often show impaired cerebrovascular reactivity that could affect cerebral perfusion [38,146,147].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10020439
This entry is offline, you can click here to edit this entry!