Non-Hodgkin lymphoma (NHL) is the most frequent hematological neoplasm in the world with more than 544,000 new NHL cases diagnosed in 2020 (2.8% of all cancer diagnoses). Of all the NHL subtypes, the most common is diffuse large B-cell lymphoma (DLBCL), accounting for approximately 40% of lymphoma cases. DLBCL is also one of the most aggressive subtypes; 5-year survival in elderly patients does not exceed 40%. The most common first-line treatment for DLBCL is chemoimmunotherapy containing rituximab, the so-called R-CHOP regimen (rituximab, cyclophosphamide, doxorubicin, vincristine), which fails in 30–40% of patients. Although relapsed/refractory (r/r) patients receive second-line treatment or may undergo autologous stem cell transplantation, their prognosis remains poor. Years of employment of rituximab as a core of first-line treatment and recent observations on CAR-T cell therapy clearly demonstrate that the use of immunotherapy invariably leads to the induction of resistance.
- diffuse large B-cell lymphoma
- immunotherapy resistance
- antibody-dependent cellular cytotoxicity
- complement-dependent cytotoxicity
1. Diffuse Large B-Cell Lymphoma
2. Rituximab (RTX)
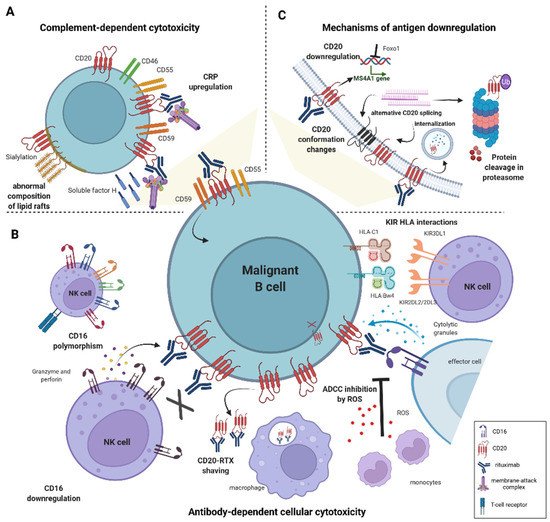
3. Mechanisms of Rituximab Cytotoxicity
This entry is adapted from the peer-reviewed paper 10.3390/ijms23031501
References
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Padala, S.A.; Barsouk, A.; Rawla, P. Epidemiology of Non-Hodgkin’s Lymphoma. Med. Sci. 2021, 9, 5.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Shaw, J.; Harvey, C.; Richards, C.; Kim, C. Temporal Trends in Treatment and Survival of Older Adult Diffuse Large B-Cell Lymphoma Patients in the SEER-Medicare Linked Database. Leuk. Lymphoma 2019, 60, 3235–3243.
- Rovira, J.; Valera, A.; Colomo, L.; Setoain, X.; Rodríguez, S.; Martínez-Trillos, A.; Giné, E.; Dlouhy, I.; Magnano, L.; Gaya, A.; et al. Prognosis of Patients with Diffuse Large B Cell Lymphoma Not Reaching Complete Response or Relapsing after Frontline Chemotherapy or Immunochemotherapy. Ann. Hematol. 2015, 94, 803–812.
- Crump, M.; Neelapu, S.S.; Farooq, U.; Van Den Neste, E.; Kuruvilla, J.; Westin, J.; Link, B.K.; Hay, A.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in Refractory Diffuse Large B-Cell Lymphoma: Results from the International SCHOLAR-1 Study. Blood 2017, 130, 1800–1808.
- Nowakowski, G.S. Recently Approved Drugs Herald a New Era in Therapy for Diffuse Large B-Cell Lymphoma. Clin. Adv. Hematol. Oncol. 2021, 19, 284–287.
- Wang, L.; Li, L.; Young, K.H. New Agents and Regimens for Diffuse Large B Cell Lymphoma. J. Hematol. Oncol. 2020, 13, 175.
- He, M.Y.; Kridel, R. Treatment Resistance in Diffuse Large B-Cell Lymphoma. Leukemia 2021, 35, 2151–2165.
- Engelhard, M. Anti-CD20 Antibody Treatment of Non-Hodgkin Lymphomas. Clin. Immunol. 2016, 172, 101–104.
- Salles, G.; Barrett, M.; Foà, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv. Ther. 2017, 34, 2232–2273.
- Winiarska, M.; Glodkowska-Mrowka, E.; Bil, J.; Golab, J. Molecular Mechanisms of the Antitumor Effects of Anti-CD20 Antibodies. Front. Biosci. (Landmark Ed) 2011, 16, 277–306.
- Johnson, P.; Glennie, M. The Mechanisms of Action of Rituximab in the Elimination of Tumor Cells. Semin. Oncol. 2003, 30, 3–8.
- Pavlasova, G.; Mraz, M. The Regulation and Function of CD20: An “Enigma” of B-Cell Biology and Targeted Therapy. Haematologica 2020, 105, 1494–1506.
- Alas, S.; Emmanouilides, C.; Bonavida, B. Inhibition of Interleukin 10 by Rituximab Results in Down-Regulation of Bcl-2 and Sensitization of B-Cell Non-Hodgkin’s Lymphoma to Apoptosis. Clin. Cancer Res. 2001, 7, 709–723.
- Jazirehi, A.R.; Bonavida, B. Cellular and Molecular Signal Transduction Pathways Modulated by Rituximab (Rituxan, Anti-CD20 MAb) in Non-Hodgkin’s Lymphoma: Implications in Chemosensitization and Therapeutic Intervention. Oncogene 2005, 24, 2121–2143.
- Vega, M.I.; Baritaki, S.; Huerta-Yepez, S.; Martinez-Paniagua, M.A.; Bonavida, B. A Potential Mechanism of Rituximab-Induced Inhibition of Tumor Growth through Its Sensitization to Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Expressing Host Cytotoxic Cells. Leuk. Lymphoma 2011, 52, 108–121.