Smooth muscle cells (SMCs), present in the media layer of blood vessels, are crucial in maintaining vascular homeostasis. Upon vascular injury, SMCs show a high degree of plasticity, undergo a change from a “contractile” to a “synthetic” phenotype, and play an essential role in the pathophysiology of diseases including atherosclerosis and restenosis. Integrins are cell surface receptors, which are involved in cell-to-cell binding and cell-to-extracellular-matrix interactions. By binding to extracellular matrix components, integrins trigger intracellular signaling and regulate several of the SMC function, including proliferation, migration, and phenotypic switching.
- integrins
- smooth muscle cell
- phenotype switching
- neointimal hyperplasia
1. Integrins: A Brief Overview
2. Role of Integrins in SMC Biology
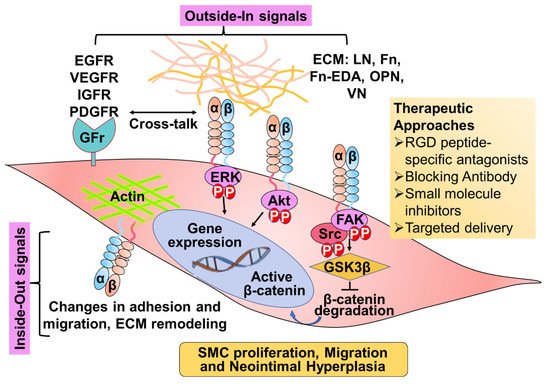
3. Role of Integrins in Neointimal Hyperplasia
| Integrin | ECM | SMC Expression | SMC Function | Implication in Atherosclerosis/Restenosis | Integrin-Targeting Agents in Clinics | Reference |
|---|---|---|---|---|---|---|
| α1β1 | Col 1-IV, LN | High expression in resting SMCs. Downregulated in culture conditions and during neointimal hyperplasia | Promotes SMC adhesion and contractile phenotype | α1β1 deletion induces a stable plaque phenotype | SAN-300 | [10,34,42,52,53,54,55] |
| α2β1 | Col 1 and IV, LN | Undetectable levels in normal human SMCs, and high expression in cultured SMCs | promote chemotaxis of arterial SMCs | α2β1 deletion had no effect on atherosclerosis | Vatelizu-mab | [16,39,42,52,56] |
| α3β1 | Col 1, Fn, and LN | Detectable levels in normal human SMCs, and high expression in cultured SMCs | No conclusive reports | [10,33] | ||
| α4β1 | Cellular-Fn, VCAM, OPN | Undetectable levels in normal human SMCs, expressed in SMCs in culture and in intimal atherosclerotic thickening | Induction of SMC differentiation | blocking α4β1 prevents neointimal hyperplasia | Natalizu-mab AJM300 |
[38,44,45,52] |
| α5β1 | Fn and LN | Low levels in normal human SMCs, and high expression in cultured SMCs and during neointimal hyperplasia | Promote SMC proliferation and migration | Mediates early atherosclerosis | Volocixi-mab ATN61 |
[41,57,58] |
| α7β1 | LN | High levels in normal SMCs, and low expression in synthetic SMC | Promotes contractile SMC phenotype | α7 deletion promotes neointimal hyperplasia | No conclusive reports | [20,51,59,60] |
| α8β1 | Fn, TN, VN | Overexpressed in SMCs that display a contractile phenotype low expression in synthetic SMC phenotype and during neointimal hyperplasia | Promotes contractile SMC phenotype. Prevents SMC proliferation and migration | α8 deletion aggravates intimal thickening | No conclusive reports | [12,35,50,61] |
| α9β1 | Fn-EDA, TN, VCAM | Expression increases in synthetic SMC phenotype | Promotes SMC proliferation, migration, and synthetic phenotype. | α9 deletion prevents NH | ASP5094 | [21,62] |
| αvβ1 | VN, Fn | Weakly expressed in normal SMCs, and upregulated in SMCs cultured on fibronectin | Inhibits contractility in SMC exposed to serum | No conclusive reports | PLN-74809 PLN-1474 |
[41,63,64] |
| αvβ3 | VN, OPN, Fn | Weakly expressed in normal SMCs, and upregulated in SMCs cultured on fibronectin and during neointimal hyperplasia | Promotes SMC adhesion, proliferation and migration | Promotes neointimal hyperplasia | LM609, Abcixi-mab (c7E3Fab; ReoPro), Vitaxin, Intetumu-mab, Cillengitide | [16,41,65,66,67,68] |
| αvβ5 | Fib, Fn, OPN VN |
highly abundant in cultured SMCs, upregulated upon vascular injury | Promotes SMC adhesion and migration | Promotes neointimal hyperplasia | LM609 Intetumu-mab |
[67,69] |
This entry is adapted from the peer-reviewed paper 10.3390/cells11040646