Adenosine is a naturally occurring purine nucleoside that regulates various physiologic functions, including inflammation and wound healing, cardiac contraction, blood vessel formation, vasodilation, learning, memory, sleep, and arousal. Adenosine is released by neurons and glial cells. Extracellular adenosine modulates neuronal excitability, synaptic plasticity, and the release and reuptake of several neurotransmitters. The effects of extracellular adenosine are modulated via four subtypes of G-protein coupled adenosine receptors (GPCRs), denoted A1, A2A, A2B, and A3. Adenosine A2A receptors (A2ARs) are broadly expressed in the brain, cardiovascular system, blood vessels, spleen, thymus, leukocytes, and lung, making them an important drug target. The therapeutic potential of targeting adenosine A2ARs is immense due to their broad expression in the body and central nervous system. The role of A2ARs in cardiovascular function, inflammation, sleep/wake behaviors, cognition, and other primary nervous system functions has been extensively studied.
1. Introduction
Adenosine is a naturally occurring purine nucleoside that regulates various physiologic functions, including inflammation and wound healing, cardiac contraction, blood vessel formation, vasodilation, learning, memory, sleep, and arousal [
1,
2,
3,
4,
5,
6,
7]. Adenosine is released by neurons and glial cells [
8]. Extracellular adenosine modulates neuronal excitability, synaptic plasticity, and the release and reuptake of several neurotransmitters [
9,
10,
11,
12]. The effects of extracellular adenosine are modulated via four subtypes of G-protein coupled adenosine receptors (GPCRs), denoted A
1, A
2A, A
2B, and A
3 [
13]. Adenosine A
2A receptors (A
2ARs) are broadly expressed in the brain, cardiovascular system, blood vessels, spleen, thymus, leukocytes, and lung, making them an important drug target [
14].
2. Adenosine and Its Receptors
Adenosine was initially recognized as a physiologic regulator of coronary vascular tone; since then, a growing body of reports indicates that adenosine regulates cellular functions through specific receptors present on the cell surface [
19,
20,
21]. Adenosine is an endogenous purine nucleoside consisting of adenine and
D-ribose, and is formed through hydrolysis of
S-adenosylhomocysteine or adenosine monophosphate [
22,
23]. Adenosine formation from
S-adenosylhomocysteine relies on the intracellular activity of the enzyme
S-adenosylhomocysteine hydrolase, which bi-directionally assures the constant occupancy of a bound adenosine concentration in the cells [
24]. Different enzymes mediate the formation of adenosine from adenosine monophosphate at both intracellular and extracellular levels.
Although adenosine does not exclusively act on synapses and is not stored in synaptic vesicles, it has a direct role in synaptic processes and the regulation of various neurotransmitters in the CNS. Nucleoside transporters mediate adenosine release and reuptake mechanisms through a concentration gradient between the intracellular and extracellular spaces. Therefore, adenosine is postulated as a modulator that affects neurotransmitter release and neuronal hyper- or depolarization and regulates glial cells [
25]. Despite the modulatory role of adenosine, neurotransmitter properties are also observed for adenosine, which is due to the presence of the adenosine-producing enzyme in synapses. Extracellular adenosine acts on neurons through specific adenosine receptors [
26].
Purinergic receptors are the natural target of purine molecules such as adenosine and adenosine triphosphate. These receptors were recognized for the first time in 1978 [
27]. Two types of purinergic receptors, P1 and P2, were subsequently identified based on their pharmacologic profile [
28]. P1 receptors recognize adenosine as a primary natural ligand and are therefore also called adenosine receptors. Each of the four types of adenosine receptors, A
1R, A
2AR, A
2BR, or A
3R, is characterized by a distinct pharmacologic profile. These receptors are members of the GPCR superfamily [
17]. A
2ARs and A
2BRs are Gs-coupled receptors, and their activation increases the activity of adenylyl cyclase, the enzyme that initiates cyclic AMP (cAMP) synthesis in the cells. A
1Rs and A
3Rs are Gi/q coupled receptors, and their activation through adenosine or agonist molecules inhibits the activity of adenylyl cyclase, which suppresses cAMP synthesis in the cells.
3. A2AR and Its Physiologic Roles
The four types of adenosine receptors, A
1R, A
2AR, A
2BR, or A
3R, react with extracellular adenosine [
13]. The activation of A
2BRs reportedly requires a high adenosine concentration. Unlike A
2BRs, adenosine levels under basal physiologic conditions are adequate to activate A
1Rs, A
2ARs, and A
3Rs with relatively equal potency. The pharmacologic strength of an endogenous ligand or agonist at its receptor, however, relies on the number of receptors on the cells. Higher concentrations of adenosine are needed to show an effect in the presence of only a few receptors. Local expression of the A
1Rs and A
2ARs in the brain is suggested to be relatively higher than that of the other two adenosine receptors [
6,
29].
A
2ARs were first identified by Libert and colleagues when they cloned several orphan GPCRs from the dog thyroid [
30]. Afterward, A
2ARs were cloned from other species, including guinea pigs, mice, rats, and humans [
31,
32,
33,
34]. As with the other GPCRs, A
2ARs induce classical secondary messenger pathways. The A
2AR signaling pathway may vary depending on the cell and tissue type in which the receptors occur. For example, Gs is the major G-protein associated with A
2ARs in the peripheral system. On the other hand, A
2ARs in the striatum, where they are highly expressed, mediate their effects mainly through Golf activation in the rat. Active Gs and Golf proteins stimulate adenylyl cyclase (
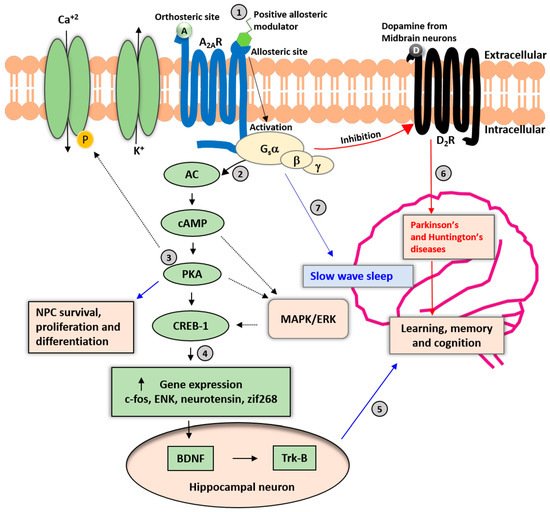
Figure 1) which increases cellular cAMP levels and activates protein kinase A (PKA) which then phosphorylates and promotes cAMP-responsive element-binding protein 1 (CREB1) [
16,
35].
Figure 1. Neuronal A2AR signaling cascades. A2AR is a Gs(olf)-protein-coupled receptor involved in various physiologic processes. (1) The allosteric modulation sites may be pharmacologically relevant for avoiding adverse effects on the cardiovascular and other peripheral systems. (2) Binding of adenosine and an allosteric modulator to A2ARs enhances the activation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA), resulting in the phosphorylation of calcium ion channels and increased influx of Ca+2 into the cytoplasm. (3) The PKA pathway also promotes neural progenitor cell (NPC) survival, proliferation, and differentiation; and activation of the mitogen-activated protein (MAP)-kinase pathway. (4) PKA-mediated phosphorylation of the cAMP-responsive element binding protein 1 (CREB-1) regulates the expression of genes such as c-fos, enkephalin (ENK), neurotensin, and zinc finger protein 268 (zif268). (5) The secretion of brain-derived neurotrophic factor (BDNF) and activation of tropomyosin receptor kinase B (TrkB) receptors in response to A2AR activation in hippocampal neurons may be relevant for cognitive functions such as learning and memory. (6) A2AR activation may be a counter mechanism to control the activation and expression of dopamine D2 receptors (D2Rs). Long-term imbalance of D2R signaling leads to impairments in cognitive and motor functions and the development of Parkinson’s and Huntington’s diseases. (7) Activation of A2AR in the nucleus accumbens increases slow-wave sleep in mice. Solid black arrows represent the primary signaling pathway of A2ARs, and dashed black arrows represent secondary signaling pathways. A: Adenosine; D: Dopamine.
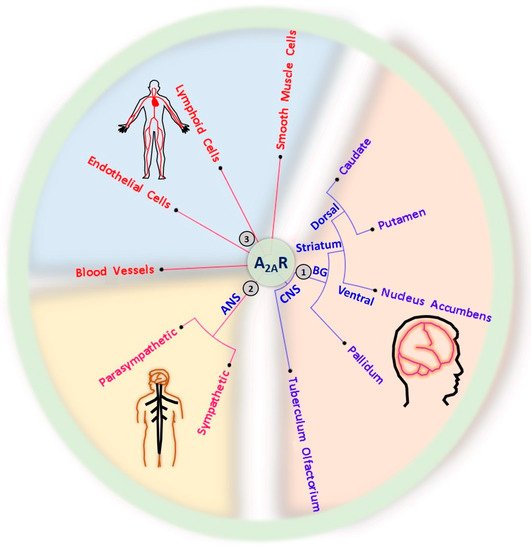
A
2ARs play a significant role in regulating the indirect pathways of the basal ganglia in the brain (
Figure 2) [
16].
Figure 2. Expression of A2ARs in the central nervous system (CNS), autonomic nervous system (ANS), circulatory system, and musculoskeletal system. (1) CNS A2ARs are mainly expressed in the basal ganglia (BG), including the dorsal pallidum, the nucleus accumbens in the ventral part of the striatum, and the dorsal striatum comprising the caudate and putamen. (2) A2ARs are also expressed in the sympathetic and parasympathetic ANS. (3) The distribution of A2ARs is not limited to the nervous system; A2ARs are also found in the circulation system, including heart, blood vessels, lymphoid cells (immune cells), and smooth muscle cells of the musculoskeletal system.
4. The Concept of Allosteric Modulation
The most common method to stimulate receptors in pharmacology and biochemistry is to target orthosteric sites with their endogenous ligand, agonists, or antagonists. On the other hand, studies show that receptor activity can be altered by small molecules that bind to an allosteric site different from the site where the endogenous ligand, agonists, or antagonists would bind [
49]. The small molecules that bind to the allosteric sites of the receptors are termed allosteric modulators. Unlike endogenous ligands, agonists, or antagonists, an allosteric modulator cannot itself activate or inactivate receptors but alters the receptor’s response to substrates that bind to orthosteric sites in two ways: (1) increase or decrease affinity, i.e., the ability of orthosteric substances to bind receptors, and (2) increase/decrease efficacy, i.e., the ability of orthosteric substances to activate receptors [
50]. Allosteric modulators reportedly change the receptor conformation, which alters the effect of the endogenous ligand, agonist, and antagonist binding [
51]. The concept of receptor modulation is not straightforward with respect to practical implementation. Allosteric modulators do not necessarily equally alter the affinity and efficacy of endogenous ligands, agonists, or antagonists of the receptors. An allosteric modulator may alter the efficacy or affinity of the endogenous ligand, but not that of the agonist or antagonist of the receptors or vice versa [
52].
The term ‘allostery’ was first used in enzymology studies in the early 1960s [
53,
54,
55]. Subsequently, allosteric modulation has been identified for all receptor superfamilies, including GPCRs, nuclear hormone receptors [
56,
57], receptor tyrosine kinases [
58,
59], and ligand/voltage-gated ion channels [
60,
61,
62,
63,
64]. The term “allosteric” began to be used increasingly in the literature, and a broad spectrum of allosteric modulators was described. Consequently, the classification of allosteric modulators was necessary to avoid possible confusion [
65,
66,
67]. Three properties are considered in the classification of allosteric modulators: (1) affinity modulation of the orthosteric ligand, (2) modulation of the signaling effect of the orthosteric ligand, and (3) direct effects of the allosteric modulator in the absence of the orthosteric ligand. Moreover, allosteric modulators are classified in terms of their effects on orthosteric ligands as positive allosteric modulators (PAM), negative allosteric modulators (NAM), or silent allosteric modulators, also known as neutral allosteric ligands [
68]. PAMs enhance the agonist/antagonist affinity and efficacy, whereas NAMs decrease orthosteric ligand affinity and efficacy. Unlike PAMs and NAMs, silent allosteric modulators do not affect the agonist or antagonist activity of orthosteric ligands, but bind to the allosteric site of the receptors and prevent PAMs or NAMs from binding to the same site, thereby inhibiting the activity of positive/negative allosteric modulators [
52]. It is important to note that activities of allosteric modulators are therefore limited by where and when the orthosteric ligand is released. Thus, in contrast to agonists or antagonists, allosteric modulators promise greater safety and fewer side effects in therapeutic applications.
5. Allosteric A2AR Modulation
Adenosine receptors are among the first known allosterically regulated GPCRs. Early studies demonstrated that amiloride and its analogs are allosteric A
2AR inhibitors [
17,
18,
69]. Subsequent studies revealed that the amiloride analog 5-(N,N-hexamethylene)-amiloride (HMA) is a potent allosteric A
2AR inhibitor. The other amiloride analogs, benzamil, 5-(N-methyl-N-isobutyl)amiloride (MIBA), 5-(N-methyl- N-guanidinocarbonyl-methyl)amiloride (MCGMA), and phenamil, were found to be more effective allosteric inhibitors than amiloride at rat A
2ARs [
17,
70]. Moreover, amiloride and its analogues do not affect the dissociation rate of the agonist [
3H]CGS21680 (3-{4-[2-({6-amino-9-[(2R,3R,4S,5S)-5-(ethylcarbamoyl)-3,4-dihydroxyoxolan-2-yl]-9H-purin-2-yl}amino) ethyl]phenyl}propanoic acid), but increase the dissociation rate of the antagonist [
3H]ZM241385 (4-(2-{[7-amino-2-(furan-2-yl)[
1,
2,
4]triazolo[1,5-a] [
1,
3,
5] triazin-5-yl]amino}ethyl)phenol) from A
2ARs [
71]. By contrast, sodium ions, for example, deteriorate the dissociation rate of the antagonist [
3H]ZM241385 from A
2ARs in a dose-dependent manner [
17]. It is important to note that other adenosine receptor agonists and antagonists are differentially affected by amilorides [
70]. A new approach specifically targeting the sodium ion pocket, known as fragment-screening based on affinity mass spectrometry, led to the discovery of fragment Fg754 as a new A
2AR NAM carrying a novel azetidine moiety and exhibiting inhibitory potency comparable to HMA. Subsequent simulations of the molecular dynamics, structure-activity relationship studies of the ligand, and nuclear magnetic resonance analyses in solution revealed the unique binding mode and antagonistic properties of Fg754, which is distinctly different from HMA [
72]. In addition, cholesterol is reported to be a weak PAM of A
2ARs [
73].
6. Allosteric A2AR Modulators and Their Potential Clinical Application
Allosteric A
2AR modulation could be a new target for drug discovery [
88]. Allosteric modulators can selectively elicit a physiologic response where and when the orthosteric ligand is released, thereby reducing the risk of an adverse effect of A
2AR activation. Moreover, the possibility of saturating allosteric effects offers greater potential for fine-tuning the physiologic response in a positive or negative direction. As allosteric modulators have no pharmacologic effect beyond the saturation dose, these molecules are associated with a lower risk for adverse effects than orthosteric ligands, giving them a potential therapeutic advantage over classical agonists and antagonists [
18,
89].
Some compounds act as allosteric A
2AR modulators, such as sodium ions, amiloride, and potassium-sparing diuretics, that also modulate other GPCRs than A
2ARs [
90]. For example, PD120918 is reported to enhance the activity of A
2AR agonists in the rat striatum [
91]. In contrast, thiadiazoles such as SCH-202676 alter the binding characteristics A
2AR agonists and antagonists [
92]. Some studies, however, suggest that thiadiazoles act as binding or oxidizing agents for SH groups rather than as allosteric modulators [
92]. To date, only a relatively small number of selective allosteric A
2AR modulators have been reported (
Table 1) [
93].
Table 1. Allosteric A2AR modulators and their functions.
|
Name
|
Type
|
Pharmacology
|
Structure
|
Physiologic Effects
|
|
3,4-Difluoro-2-((2-fluoro-4-iodophenyl)amino)benzoic acid
|
Allosteric enhancer/modulator
|
Enhanced adenosine signaling at mouse A2ARs.
|
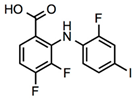
|
Induced slow wave sleep without affecting cardiovascular function or body temperature in wild-type male mice [94,95].
|
|
AEA061
|
Allosteric enhancer/modulator
|
Enhanced adenosine and inosine signaling and increased effect of the A2AR agonist CGS 21680.
|
Not disclosed
|
Inhibited the production of tumor necrosis factor-α, macrophage inflammatory protein-1α, 1β, and 2, interleukin-1α, keratinocyte chemokine, and RANTES (regulated upon activation, normal T cell expressed and presumably secreted) in macrophages and splenocytes, reduced circulating plasma tumor necrosis factor-α and monocyte chemoattractant protein-1 levels, and increased plasma interleukin-10 during lipopolysaccharide-induced endotoxemia [96,97].
|
|
N-(3-Benzyl-5-phenyl-3H-[1,2,3]triazolo[4,5-d]- pyrimidin-7yl-)-(4-aminophenyl)-amine
|
Allosteric modulator
|
Inhibited the binding of antagonists and agonists at the A2AR orthosteric site [93].
|
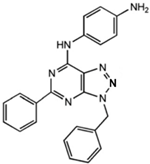
|
Unknown
|
|
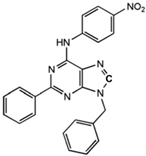
N6-[(4-Nitro)-phenyl]-9-benzyl-2-phenyladenine
|
Allosteric modulator
|
Inhibited the binding of antagonists and agonists at the A2AR orthosteric site [93].
|
|
Unknown
|
|
N6-[(4-Amino)-phenyl]-9-benzyl-2-phenyladenine
|
Allosteric modulator
|
Inhibited the binding of antagonists and agonists at the A2AR orthosteric site [93].
|
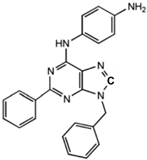
|
Unknown
|
|
1-[4-(3-Benzyl-5-phenyl-3H-[1,2,3]triazolo[4,5-d]-pyrimidin-7-ylamino)-phenyl]-3-(4-fluorophenyl)-urea
|
Allosteric modulator
|
Modulated the binding of antagonist and agonist at the A2AR orthosteric site [93].
|
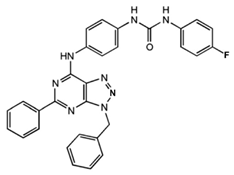
|
Unknown
|
|
1-[4-(3-Benzyl-5-phenyl-3H-[1,2,3]triazolo[4,5-d]-pyrimidin-7-ylamino)-phenyl]-3-(4-trifluoromethylphenyl)- urea
|
Allosteric modulator
|
Modulated the binding of antagonist and agonist at the A2AR orthosteric site [93].
|
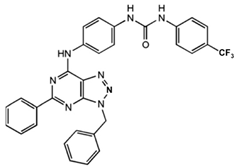
|
Unknown
|
|
1-[4-(9-Benzyl-2-phenyl-9H-purin-6-ylamino)- phenyl]-3-(4-methoxyphenyl-urea
|
Allosteric modulator
|
Modulated the binding of antagonist and agonist at the A2AR orthosteric site [93].
|
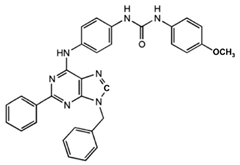
|
Unknown
|
|
Amiloride
|
Allosteric modulator
|
Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [18,71].
|
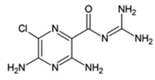
|
Unknown
|
|
Benzamil
|
Allosteric modulator
|
Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71].
|
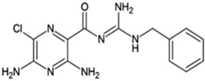
|
Unknown
|
|
HMA; 5-(N,N-hexamethylene)amiloride
|
Allosteric modulator
|
Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71].
|
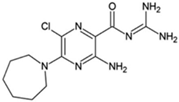
|
Unknown
|
|
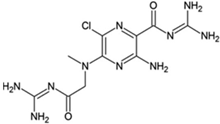
MGCMA; 5-(N-methyl-N-guanidinocarbonyl-methyl)amiloride
|
Allosteric modulator
|
Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71].
|
|
Unknown
|
|
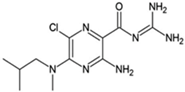
MIBA; 5-(N-methyl-N-isobutyl)amiloride
|
Allosteric modulator
|
Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71].
|
|
Unknown
|
|
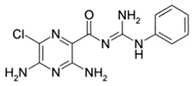
Phenamil
|
Allosteric modulator
|
Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71].
|
|
Unknown
|
|
Sodium Ion
|
Allosteric modulator
|
Positively modulated A2ARs [71].
|
Na+
|
Unknown
|
|
PD120918 {4-methyl-7-[(methyl- amino)carbonyl]oxy}-2H-1-benzopyran-2-one}
|
Allosteric modulator
|
Enhanced agonist radioligand binding to rat striatal A2ARs without functional enhancement [18,91].
|
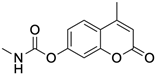
|
Unknown
|
|
Fg754
|
Allosteric modulator
|
Increased the dissociation rate of the agonist CGS21680 at A2ARs expressing HEK-293 cells [72].
|
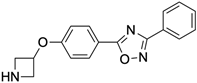
|
Unknown
|
|
Cholesterol
|
Allosteric modulator
|
Decreased the dissociation rate of the agonist NECA at A2ARs-embedded nanodiscs [73].
|
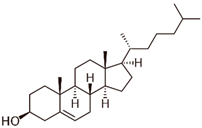
|
Unknown
|
This entry is adapted from the peer-reviewed paper 10.3390/ijms23042101