Amyloidosis is a clinical and pathological condition in which amyloid accumulates in various organs and cells of the body, forming amyloid plaques for complex reasons, leading to organ dysfunction. It can be hereditary or acquired. Depending on the location of amyloid fibers’ deposition, amyloidosis is divided into two groups, one is localized amyloidosis, that occurs in a specific area of a single tissue, and the other is systemic amyloidosis, which occurs throughout the body. Amyloid plaques are made up of amyloid proteins. Amyloid β peptide (Aβ) is the main component that plays a significant role in the pathogenesis of AD and is considered to be the leading cause of Alzheimer’s disease (AD) development.
- Alzheimer’s disease
- amyloidosis
1. Role of Amyloidosis and Its Precursor in Alzheimer’s Disease (AD)
1.1. Amyloidosis
1.2. Amyloid Precursor Protein
1.3. Amyloid Beta Protein
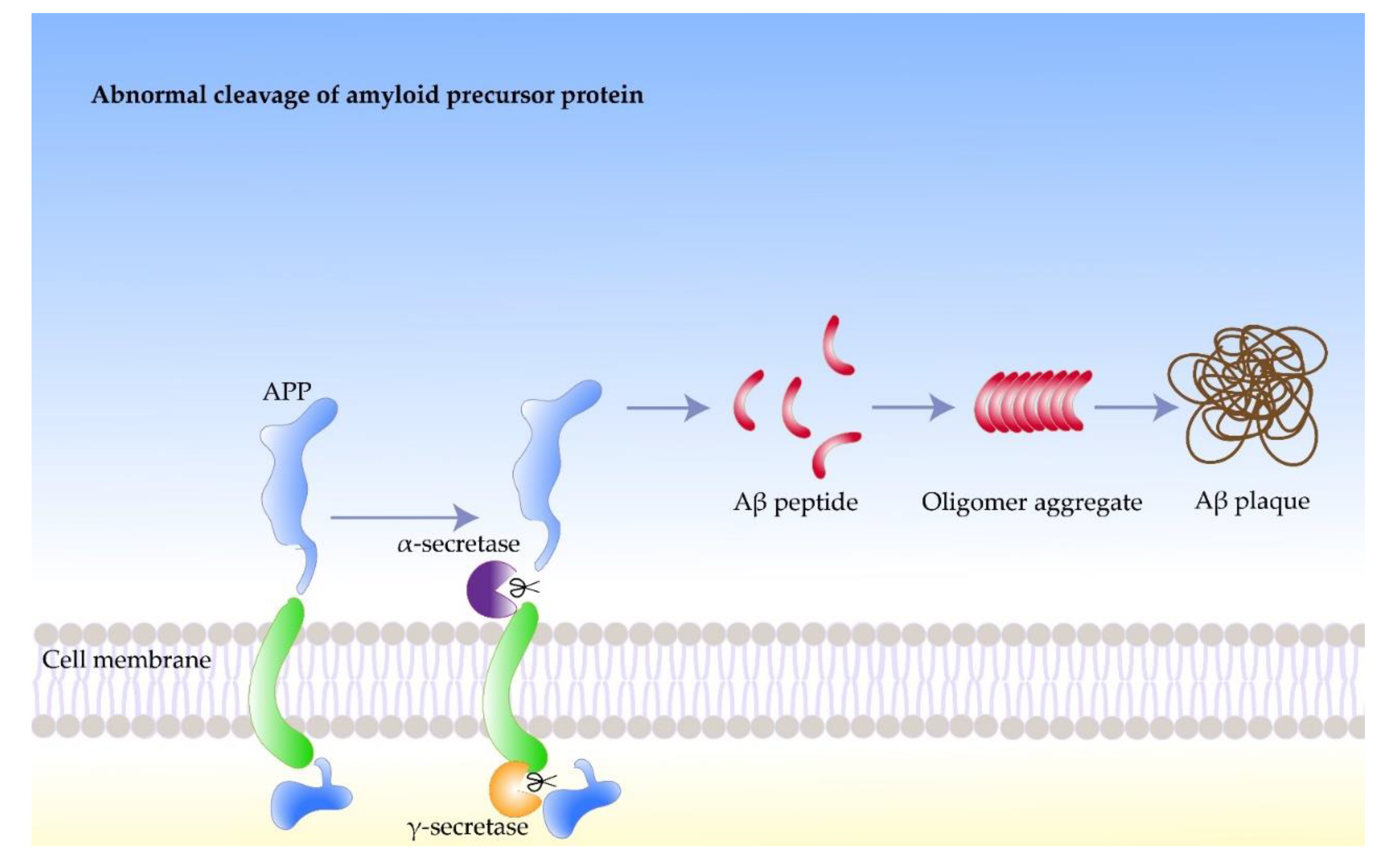
2. Inducing Factors of Amyloidosis in AD
2.1. Impaired Autophagy
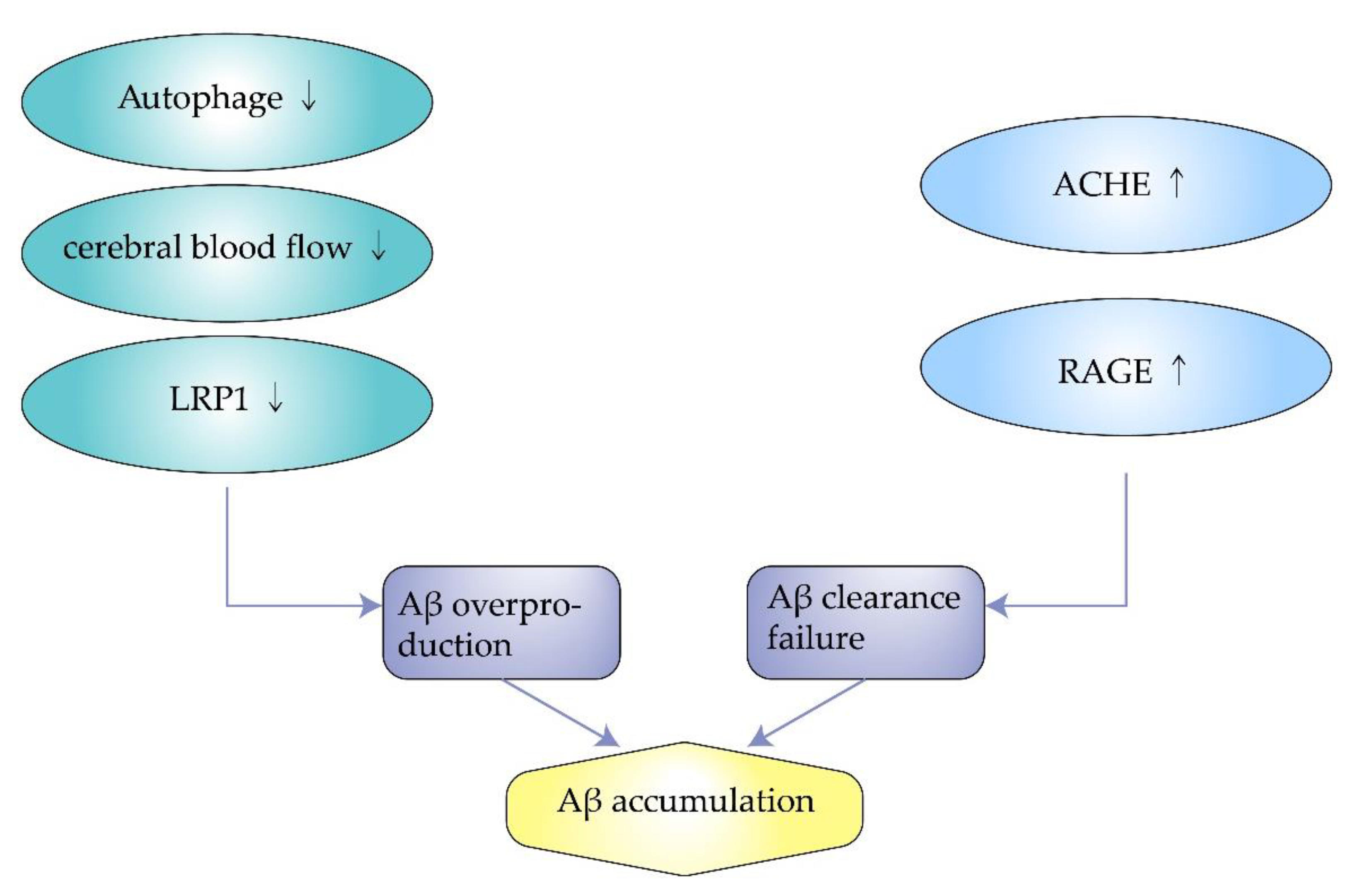
2.2. Insufficient Cerebral Blood Flow
2.3. Excess Synthesis of ACHE
2.4. Decreased Expression of Low-Density Lipoprotein Receptor-Related Protein-1
2.5. Overexpression of Receptor for Advanced Glycosylation End Products
3. Pathogenesis of Amyloidosis in AD
3.1. Oxidative Damage
3.2. Aberrant Phosphorylation of Tau
3.3. Neurofibrillary Tangles
3.4. Neuronal Damage
4. Therapeutic Directions of Amyloidosis in AD
4.1. Inhibition of Aβ Production
4.2. Reduction in Aβ Deposition
4.3. Protection of Neurons
This entry is adapted from the peer-reviewed paper 10.3390/molecules27041210
References
- Westermark, P.; Benson, M.D.; Buxbaum, J.N.; Cohen, A.S.; Frangione, B.; Ikeda, S.; Masters, C.L.; Merlini, G.; Saraiva, M.J.; Sipe, J.D. A primer of amyloid nomenclature. Amyloid 2007, 14, 179–183.
- Prasansuklab, A.; Tencomnao, T. Amyloidosis in Alzheimer’s Disease: The Toxicity of Amyloid Beta (Aβ), Mechanisms of Its Accumulation and Implications of Medicinal Plants for Therapy. Evid. Based Complement. Alternat. Med. 2013, 2013, 413808.
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. 1984. Biochem. Biophys. Res. Commun. 2012, 425, 534–539.
- Merlini, G.; Bellotti, V. Molecular mechanisms of amyloidosis. N. Engl. J. Med. 2003, 349, 583–596.
- Rice, H.C.; de Malmazet, D.; Schreurs, A.; Frere, S.; Van Molle, I.; Volkov, A.N.; Creemers, E.; Vertkin, I.; Nys, J.; Ranaivoson, F.M.; et al. Secreted amyloid-beta precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science 2019, 363.
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204.
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619.
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403.
- Sadleir, K.R.; Kandalepas, P.C.; Buggia-Prevot, V.; Nicholson, D.A.; Thinakaran, G.; Vassar, R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 235–256.
- Nalivaeva, N.N.; Turner, A.J. The amyloid precursor protein: A biochemical enigma in brain development, function and disease. FEBS Lett. 2013, 587, 2046–2054.
- Storey, E.; Cappai, R. The amyloid precursor protein of Alzheimer’s disease and the Abeta peptide. Neuropathol. Appl. Neurobiol. 1999, 25, 81–97.
- Rogaev, E.I. Genetic factors and a polygenic model of Alzheimer’s disease. Genetika 1999, 35, 1558–1571.
- Devkota, S.; Williams, T.D.; Wolfe, M.S. Familial Alzheimer’s disease mutations in amyloid protein precursor alter proteolysis by gamma-secretase to increase amyloid beta-peptides of >/=45 residues. J. Biol. Chem. 2021, 296, 100281.
- Selkoe, D.J. Clearing the brain’s amyloid cobwebs. Neuron 2001, 32, 177–180.
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913.
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30.
- Levin, O.S.; Vasenina, E.E. Twenty-five years of the amyloid hypothesis of alzheimer disease: Advances, failures and new perspectives. Zh. Nevrol. Psikhiatr. Im. SS Korsakova 2016, 116, 3–9.
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562.
- Fukumoto, H.; Tomita, T.; Matsunaga, H.; Ishibashi, Y.; Saido, T.C.; Iwatsubo, T. Primary cultures of neuronal and non-neuronal rat brain cells secrete similar proportions of amyloid beta peptides ending at A beta40 and A beta42. Neuroreport 1999, 10, 2965–2969.
- Tsitsopoulos, P.P.; Marklund, N. Amyloid-beta Peptides and Tau Protein as Biomarkers in Cerebrospinal and Interstitial Fluid Following Traumatic Brain Injury: A Review of Experimental and Clinical Studies. Front. Neurol. 2013, 4, 79.
- Maltsev, A.V.; Santockyte, R.; Bystryak, S.; Galzitskaya, O.V. Activation of neuronal defense mechanisms in response to pathogenic factors triggering induction of amyloidosis in Alzheimer’s disease. J. Alzheimers Dis. 2014, 40, 19–32.
- Zhou, F.; van Laar, T.; Huang, H.; Zhang, L. APP and APLP1 are degraded through autophagy in response to proteasome inhibition in neuronal cells. Protein. Cell 2011, 2, 377–383.
- Nixon, R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091.
- Hung, S.Y.; Huang, W.P.; Liou, H.C.; Fu, W.M. Autophagy protects neuron from Abeta-induced cytotoxicity. Autophagy 2009, 5, 502–510.
- Wu, S.; Wei, Y.; Li, J.; Bai, Y.; Yin, P.; Wang, S. SIRT5 Represses Neurotrophic Pathways and Abeta Production in Alzheimer’s Disease by Targeting Autophagy. ACS Chem. Neurosci. 2021, 12, 4428–4437.
- Li, D.; Liu, Y.; Zeng, X.; Xiong, Z.; Yao, Y.; Liang, D.; Qu, H.; Xiang, H.; Yang, Z.; Nie, L.; et al. Quantitative Study of the Changes in Cerebral Blood Flow and Iron Deposition During Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2020, 78, 439–452.
- Fazlollahi, A.; Calamante, F.; Liang, X.; Bourgeat, P.; Raniga, P.; Dore, V.; Fripp, J.; Ames, D.; Masters, C.L.; Rowe, C.C.; et al. Increased cerebral blood flow with increased amyloid burden in the preclinical phase of alzheimer’s disease. J. Magn. Reson. Imaging 2020, 51, 505–513.
- Winchester, J.; Dick, M.B.; Gillen, D.; Reed, B.; Miller, B.; Tinklenberg, J.; Mungas, D.; Chui, H.; Galasko, D.; Hewett, L.; et al. Walking stabilizes cognitive functioning in Alzheimer’s disease (AD) across one year. Arch. Gerontol. Geriatr. 2013, 56, 96–103.
- Maesako, M.; Uemura, K.; Kubota, M.; Kuzuya, A.; Sasaki, K.; Hayashida, N.; Asada-Utsugi, M.; Watanabe, K.; Uemura, M.; Kihara, T.; et al. Exercise is more effective than diet control in preventing high fat diet-induced beta-amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. J. Biol. Chem. 2012, 287, 23024–23033.
- Intlekofer, K.A.; Cotman, C.W. Exercise counteracts declining hippocampal function in aging and Alzheimer’s disease. Neurobiol. Dis. 2013, 57, 47–55.
- Hashiguchi, D.; Campos, H.C.; Wuo-Silva, R.; Faber, J.; Gomes da Silva, S.; Coppi, A.A.; Arida, R.M.; Longo, B.M. Resistance Exercise Decreases Amyloid Load and Modulates Inflammatory Responses in the APP/PS1 Mouse Model for Alzheimer’s Disease. J. Alzheimers Dis. 2020, 73, 1525–1539.
- Dougherty, J.J.; Wu, J.; Nichols, R.A. Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J. Neurosci. 2003, 23, 6740–6747.
- Dineley, K.T.; Bell, K.A.; Bui, D.; Sweatt, J.D. beta-Amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J. Biol. Chem. 2002, 277, 25056–25061.
- Govoni, S.; Mura, E.; Preda, S.; Racchi, M.; Lanni, C.; Grilli, M.; Zappettini, S.; Salamone, A.; Olivero, G.; Pittaluga, A.; et al. Dangerous liaisons between beta-amyloid and cholinergic neurotransmission. Curr. Pharm. Des. 2014, 20, 2525–2538.
- Marco-Contelles, J.; Unzeta, M.; Bolea, I.; Esteban, G.; Ramsay, R.R.; Romero, A.; Martinez-Murillo, R.; Carreiras, M.C.; Ismaili, L. ASS234, As a New Multi-Target Directed Propargylamine for Alzheimer’s Disease Therapy. Front. Neurosci. 2016, 10, 294.
- Simoni, E.; Bartolini, M.; Abu, I.F.; Blockley, A.; Gotti, C.; Bottegoni, G.; Caporaso, R.; Bergamini, C.; Andrisano, V.; Cavalli, A.; et al. Multitarget drug design strategy in Alzheimer’s disease: Focus on cholinergic transmission and amyloid-beta aggregation. Future Med. Chem. 2017, 9, 953–963.
- Donahue, J.E.; Flaherty, S.L.; Johanson, C.E.; Duncan, J.A., 3rd; Silverberg, G.D.; Miller, M.C.; Tavares, R.; Yang, W.; Wu, Q.; Sabo, E.; et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006, 112, 405–415.
- Sagare, A.; Deane, R.; Bell, R.D.; Johnson, B.; Hamm, K.; Pendu, R.; Marky, A.; Lenting, P.J.; Wu, Z.; Zarcone, T.; et al. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat. Med. 2007, 13, 1029–1031.
- Choi, J.; Gao, J.; Kim, J.; Hong, C.; Kim, J.; Tontonoz, P. The E3 ubiquitin ligase Idol controls brain LDL receptor expression, ApoE clearance, and Abeta amyloidosis. Sci. Transl. Med. 2015, 7, 314ra184.
- Fuentealba, R.A.; Liu, Q.; Zhang, J.; Kanekiyo, T.; Hu, X.; Lee, J.M.; LaDu, M.J.; Bu, G. Low-density lipoprotein receptor-related protein 1 (LRP1) mediates neuronal Abeta42 uptake and lysosomal trafficking. PLoS ONE 2010, 5, e11884.
- Zhou, R.; Chen, L.L.; Yang, H.; Li, L.; Liu, J.; Chen, L.; Hong, W.J.; Wang, C.G.; Ma, J.J.; Huang, J.; et al. Effect of High Cholesterol Regulation of LRP1 and RAGE on Abeta Transport Across the Blood-Brain Barrier in Alzheimer’s Disease. Curr. Alzheimer Res. 2021, 18, 428–442.
- Fang, F.; Yu, Q.; Arancio, O.; Chen, D.; Gore, S.S.; Yan, S.S.; Yan, S.F. RAGE mediates Abeta accumulation in a mouse model of Alzheimer’s disease via modulation of beta- and gamma-secretase activity. Hum. Mol. Genet. 2018, 27, 1002–1014.
- Huang, Y.Y.; Fang, N.; Luo, H.R.; Gao, F.; Zou, Y.; Zhou, L.L.; Zeng, Q.P.; Fang, S.S.; Xiao, F.; Zheng, Q. RP1, a RAGE antagonist peptide, can improve memory impairment and reduce Abeta plaque load in the APP/PS1 mouse model of Alzheimer’s disease. Neuropharmacology 2020, 180, 108304.
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658.
- Sotolongo, K.; Ghiso, J.; Rostagno, A. Nrf2 activation through the PI3K/GSK-3 axis protects neuronal cells from Abeta-mediated oxidative and metabolic damage. Alzheimers Res. Ther. 2020, 12, 13.
- Butterfield, D.A. The 2013 SFRBM discovery award: Selected discoveries from the butterfield laboratory of oxidative stress and its sequela in brain in cognitive disorders exemplified by Alzheimer disease and chemotherapy induced cognitive impairment. Free Radic. Biol. Med. 2014, 74, 157–174.
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 924–929.
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002, 32, 1050–1060.
- Malkov, A.; Popova, I.; Ivanov, A.; Jang, S.S.; Yoon, S.Y.; Osypov, A.; Huang, Y.; Zilberter, Y.; Zilberter, M. Abeta initiates brain hypometabolism, network dysfunction and behavioral abnormalities via NOX2-induced oxidative stress in mice. Commun. Biol. 2021, 4, 1054.
- Kapasi, A.; Leurgans, S.E.; Arvanitakis, Z.; Barnes, L.L.; Bennett, D.A.; Schneider, J.A. Abeta (Amyloid Beta) and Tau Tangle Pathology Modifies the Association Between Small Vessel Disease and Cortical Microinfarcts. Stroke 2021, 52, 1012–1021.
- Hurtado, D.E.; Molina-Porcel, L.; Iba, M.; Aboagye, A.K.; Paul, S.M.; Trojanowski, J.Q.; Lee, V.M. A accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am. J. Pathol. 2010, 177, 1977–1988.
- Wu, H.; Wei, S.; Huang, Y.; Chen, L.; Wang, Y.; Wu, X.; Zhang, Z.; Pei, Y.; Wang, D. Abeta monomer induces phosphorylation of Tau at Ser-214 through beta2AR-PKA-JNK signaling pathway. FASEB J. 2020, 34, 5092–5105.
- Gilley, J.; Ando, K.; Seereeram, A.; Rodriguez-Martin, T.; Pooler, A.M.; Sturdee, L.; Anderton, B.H.; Brion, J.P.; Hanger, D.P.; Coleman, M.P. Mislocalization of neuronal tau in the absence of tangle pathology in phosphomutant tau knockin mice. Neurobiol. Aging 2016, 39, 1–18.
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol. Neurodegener. 2021, 16, 37.
- Minati, L.; Edginton, T.; Bruzzone, M.G.; Giaccone, G. Current concepts in Alzheimer’s disease: A multidisciplinary review. Am. J. Alzheimers Dis. Other Demen. 2009, 24, 95–121.
- Goedert, M. Neurodegeneration. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 2015, 349, 1255555.
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539.
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842.
- Parodi, J.; Sepulveda, F.J.; Roa, J.; Opazo, C.; Inestrosa, N.C.; Aguayo, L.G. Beta-amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J. Biol. Chem. 2010, 285, 2506–2514.
- Sepulveda, F.J.; Parodi, J.; Peoples, R.W.; Opazo, C.; Aguayo, L.G. Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLoS ONE 2010, 5, e11820.
- Peters, C.; Fernandez-Perez, E.J.; Burgos, C.F.; Espinoza, M.P.; Castillo, C.; Urrutia, J.C.; Streltsov, V.A.; Opazo, C.; Aguayo, L.G. Inhibition of amyloid beta-induced synaptotoxicity by a pentapeptide derived from the glycine zipper region of the neurotoxic peptide. Neurobiol. Aging 2013, 34, 2805–2814.
- Du, H.; Guo, L.; Yan, S.S. Synaptic mitochondrial pathology in Alzheimer’s disease. Antioxid. Redox Signal. 2012, 16, 1467–1475.
- Schelle, J.; Wegenast-Braun, B.M.; Fritschi, S.K.; Kaeser, S.A.; Jährling, N.; Eicke, D.; Skodras, A.; Beschorner, N.; Obermueller, U.; Häsler, L.M.; et al. Early Aβ reduction prevents progression of cerebral amyloid angiopathy. Ann. Neurol. 2019, 86, 561–571.
- Baranger, K.; Giannoni, P.; Girard, S.D.; Girot, S.; Gaven, F.; Stephan, D.; Migliorati, M.; Khrestchatisky, M.; Bockaert, J.; Marchetti-Gauthier, E.; et al. Chronic treatments with a 5-HT(4) receptor agonist decrease amyloid pathology in the entorhinal cortex and learning and memory deficits in the 5xFAD mouse model of Alzheimer’s disease. Neuropharmacology 2017, 126, 128–141.
- Du, Y.; Qu, J.; Zhang, W.; Bai, M.; Zhou, Q.; Zhang, Z.; Li, Z.; Miao, J. Morin reverses neuropathological and cognitive impairments in APPswe/PS1dE9 mice by targeting multiple pathogenic mechanisms. Neuropharmacology 2016, 108, 1–13.
- Zhao, C.; Zhang, H.; Li, H.; Lv, C.; Liu, X.; Li, Z.; Xin, W.; Wang, Y.; Zhang, W. Geniposide ameliorates cognitive deficits by attenuating the cholinergic defect and amyloidosis in middle-aged Alzheimer model mice. Neuropharmacology 2017, 116, 18–29.
- Esquerda-Canals, G.; Roda, A.R.; Marti-Clua, J.; Montoliu-Gaya, L.; Rivera-Hernandez, G.; Villegas, S. Treatment with scFv-h3D6 Prevented Neuronal Loss and Improved Spatial Memory in Young 3xTg-AD Mice by Reducing the Intracellular Amyloid-beta Burden. J. Alzheimers Dis. 2019, 70, 1069–1091.
- Yang, C.; Mo, Y.S.; Chen, H.F.; Huang, Y.H.; Li, S.L.; Wang, H.; Huang, S.Q.; Chang, X.; Du, Q.; Wang, Q. The effects of Danggui-Shaoyao-San on neuronal degeneration and amyloidosis in mouse and its molecular mechanism for the treatment of Alzheimer’s disease. J. Integr. Neurosci. 2021, 20, 255–264.
- Chen, M.; Li, L.; Liu, C.; Song, L. Berberine attenuates Abeta-induced neuronal damage through regulating miR-188/NOS1 in Alzheimer’s disease. Mol. Cell Biochem. 2020, 474, 285–294.
- Maiti, P.; Bowers, Z.; Bourcier-Schultz, A.; Morse, J.; Dunbar, G.L. Preservation of dendritic spine morphology and postsynaptic signaling markers after treatment with solid lipid curcumin particles in the 5xFAD mouse model of Alzheimer’s amyloidosis. Alzheimers Res. Ther. 2021, 13, 37.
- Xiao, H.; Li, H.; Song, H.; Kong, L.; Yan, X.; Li, Y.; Deng, Y.; Tai, H.; Wu, Y.; Ni, Y.; et al. Shenzao jiannao oral liquid, an herbal formula, ameliorates cognitive impairments by rescuing neuronal death and triggering endogenous neurogenesis in AD-like mice induced by a combination of Abeta42 and scopolamine. J. Ethnopharmacol. 2020, 259, 112957.