Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Urology & Nephrology
Hyperfiltration is an important underlying cause of glomerular dysfunction associated with several systemic and intrinsic glomerular conditions leading to chronic kidney disease (CKD). These include obesity, diabetes, hypertension, focal segmental glomerulosclerosis (FSGS), congenital abnormalities and reduced renal mass (low nephron number). Hyperfiltration-associated biomechanical forces directly impact the cell membrane, generating tensile and fluid flow shear stresses in multiple segments of the nephron.
- hyperfiltration
1. Introduction
1.1. Glomerular Hyperfiltration Is an Early Response That May Turn Maladaptive
Plasma filtration in the glomerulus and ultrafiltrate processing in the tubular segment are primary physiological functions of the nephron. Capillary endothelial cells, mesangial cells, glomerular basement membrane (GBM) and podocytes interact to constitute the glomerular filtration barrier modulated by multiple hemodynamic, neurohumoral and immune factors. Consistent glomerular filtration is essential for maintaining homeostasis and estimated glomerular filtration rate (eGFR) is an important clinical parameter for evaluating kidney function. Estimated GFR between 120–90 mL/min/1.73 m2 reflecting a physiological decline with age is considered normal for healthy adults (20–70 years). However, eGFR below 60 mL/min/1.73 m2 indicates chronic kidney disease (CKD) with moderate (59–30 mL, Stage 3) to severe (29–15 mL, Stage 4) loss of kidney function and kidney failure (Stage 5 CKD) below 15 mL/min/1.73 m2 [1,2].
Higher eGFR reflects an increased rate of glomerular filtration, termed glomerular hyperfiltration (GHF). A precise definition, threshold levels/range and an exact mechanism of GHF are subjects of ongoing studies and discussion. GHF is variously defined based on high whole kidney GFR, elevated filtration fraction (GFR/Renal blood flow *100) or increased single nephron glomerular filtration rate (SNGFR) to describe total renal filtration capacity. In large population studies, eGFR above the 95th or 97th percentile of the cohort is also used as the threshold value for GHF. A median threshold value of 135 mL/min/1.73 m2 (range 90.7–175 mL, most values between 135 and 140 mL) has been reported [3]. A recent study on diabetes defined GHF at GFR ≥140 mL/min per 1.73 m2, with secondary thresholds of 130 or 150 mL/min per 1.73 m2 [4]. Others have used 120 mL/min/1.73 m2 as the cutoff for GHF [5] which is closer to the upper limit of eGFR in healthy adults (90–120 mL).
An adaptive increase in GFR during pregnancy occurs due to physiological endocrine changes that result in higher cardiac output and lower peripheral resistance causing increased renal blood flow and glomerular size [6,7]. A transient physiological increase in GFR also occurs after consuming high-protein meals. Long-term high protein consumption may cause adverse renal effects in vulnerable groups such as obese individuals with subclinical renal damage [8,9,10]. Initial adaptive increase in GFR causes transient hemodynamic changes. However, adaptive changes may turn maladaptive and pathological due to persistent insult. Glomerular hyperfiltration followed by a gradual decline in filtration parallel structural changes in the glomerular and tubular compartment are outlined in Figure 1.
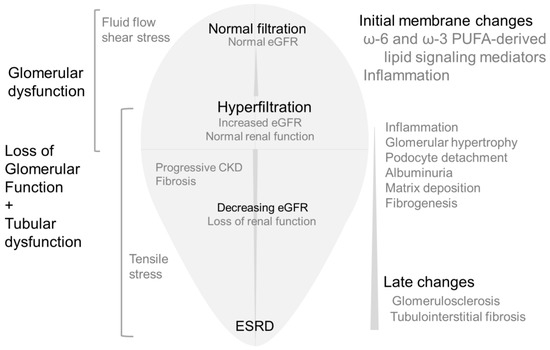
Figure 1. Simplified version of renal changes caused by hyperfiltration followed by lower filtration.Middle: Initially, glomerular filtration rate increases (hyperfiltration), followed by a decrease in GFR and CKD leading to ESRD. Left: Increased fluid flow shear stress drives effects of hyperfiltration in the early stages indicated by glomerular/podocyte dysfunction. Gradual increase in tensile stress is associated with rapid loss of glomerular function and tubular changes causing CKD. Right: Cellular stress causes inflammatory changes and the release of fatty acids from membrane phospholipids. Fatty acid metabolites mediate mechanotransduction and activate cellular signaling pathways as an initial response to hyperfiltration. With time, tubular homeostasis also changes in response to early glomerular changes. Initial lipid-mediated signaling events are followed by more complex and diverse signaling and functional changes resulting in albuminuria, matrix accumulation, fibrogenesis, podocyte loss leading to glomerulosclerosis and fibrosis.
1.2. Glomerular Hyperfiltration Is Associated with Several Pathophysiological Etiologies
Maladaptive GHF, while not unique to a specific condition, is an underlying risk factor of early glomerular dysfunction in several diseases where normal kidneys are vulnerable to systemic or primary glomerular pathophysiological changes. GHF has been mostly described in the context of the early stages of obesity, diabetes and hypertension that are components of the metabolic syndrome spectrum which represents a co-occurrence of metabolic risk factors (abdominal obesity, hyperglycemia, dyslipidemia and hypertension) for type 2 diabetes and cardiovascular disease (CVD) [11,12]. GHF in humans may also be associated with hyperperfusion independent of blood pressure or diabetes [13]. GHF has been described in several other diseases that range from non-alcoholic fatty liver disease (NAFLD) to dementia and stroke. A lower nephron number due to unilateral renal agenesis or reduced functional renal mass due to other congenital abnormalities cause hyperfiltration early in life. Kidney donation also results in hyperfiltration leading to CKD after 20+ years in 3–10% of donors [14,15,16,17]. The growing number of studies involving subjects with single functional kidneys and relevant animal models of hyperfiltration will add valuable information to diabetes, obesity, and hypertension studies. Table 1 provides a summary of the conditions associated with GHF. Each of these conditions, with its unique features and variations, is a subject of detailed studies, some of which are cited.
Table 1. Hyperfiltration is an early event in several kidney diseases.
| Pathophysiology Associated with Hyperfiltration/Kidney Disease | References |
|---|---|
| High dietary protein consumption by vulnerable groups | [18,19,20] |
| Obesity | [21,22,23,24,25,26,27,28,29] |
| Diabetes | [30,31,32,33,34,35,36,37,38,39,40,41,42,43,44] |
| Hypertension | [45,46,47,48,49,50,51,52] |
| Primary hyperaldosteronism | [53,54,55,56] |
| Non-alcoholic fatty liver disease (NAFLD) | [57,58,59,60,61,62] |
| CKD in Kidney donors | [14,17,63,64,65,66,67,68] |
| CKD in Children born with single functioning kidney or low number of functional nephrons due to other Congenital Anomalies of the Kidney and Urinary Tract (CAKUT) | [16,69,70,71,72,73] |
| Autosomal Dominant Polycystic Kidney Disease (ADPKD) | [74] |
| Secondary focal segmental glomerulosclerosis (FSGS) | [75,76,77,78] |
| Sickle cell Disease (SCD) and glomerular sclerosis | [79,80,81,82] |
| Cyanotic congenital heart disease/critical congenital heart disease (CCHD) | [83,84,85,86,87] |
| ‘Autoimmune activation’ and inflammation | [88,89] |
| High altitude renal syndrome | [90] |
| Dementia | [91] |
| Stroke | [92,93] |
1.3. Glomerular Hyperfiltration Is a Potential Predictor of CKD and Cardiovascular Disease
In addition to decreased eGFR, elevated blood pressure and microalbuminuria are clinically used indicators of kidney damage and decreased function. Albuminuria is the benchmark indicator of glomerular dysfunction, and it directly exacerbates tubular dysfunction due to oxidative stress, thus adding to the deterioration of renal function. Proteinuria at >1 g/day is prognostic of progressive renal disease, cardiovascular disease (CVD), and poor outcome. Nevertheless, these indicators represent existing low renal function and at least minimum structural change.
Increased glomerular filtration is an adaptive response mounted during the early stages, prior to functional loss. GHF is considered a surrogate marker of elevated intraglomerular pressure in patients with diabetes mellitus [94] and predictor of CKD [27] and it associates with increased risk of cardiovascular disease and all-cause mortality [95,96,97]. However, a lack of detailed understanding of early hyperfiltration and its mechanism has perhaps hindered the use of hyperfiltration as a clinical indicator of renal dysfunction.
1.4. Glomerular Hyperfiltration Precedes Tissue Fibrosis and Organ Failure
Hyperfiltration and renal fibrosis are temporally distinct events in the development and progression of CKD. Cellular stress caused by hyperfiltration induces changes in the plasma membrane, leading to signaling for mechanotransduction. These early events initiate increasingly complex responses that lead to fibrogenesis and end-stage renal disease (ESRD). Early events also trigger the synthesis of matrix proteins that begin to accumulate in the absence of matched degradation by metalloproteases. Continued matrix accumulation triggers uncontrolled and irreversible pathophysiological changes [98,99,100,101].
Initial signaling events associated with GHF are mediated mainly by membrane lipids, while subsequent fibrogenesis stimulates and links multiple signaling molecules. Inflammation and TGF-β/Smad signaling play a central role in fibrogenesis. Other key molecules and pathways related to fibrogenesis include fibroblast growth factor (FGF), platelet-derived growth factor, IL-1, TNF-α, renin-angiotensin- aldosterone system (RAAS), microRNA clusters, vitamin D, bradykinin, parathyroid hormone, eNOS and JAK/STAT pathway. Altered expression of membrane glycosaminoglycans (heparan sulfate, hyaluronic acid, etc.) also add to fibrogenesis by influencing the interactions between various effector molecules and cell receptors [102,103,104]. Thus, early intervention during hyperfiltration may slow down/prevent fibrogenesis.
2. Hyperfiltration and Biomechanical Forces
Figure 1 summarizes the temporal changes in renal structure and function caused by the hyperfiltration-induced increase in biomechanical forces. Hyperfiltration results from changes in the physicochemical characteristics of the filtration barrier due to increased afferent arteriolar dilation or efferent arteriolar constriction. These changes in the capillary are generally modeled assuming the capillary as a thin curved cylinder. This model provides a calculated difference in hydrostatic pressure at 40 mm Hg between glomerular capillary pressure (~55 mm Hg) and Bowman space (~15 mm Hg). Elevated capillary pressure increases biomechanical forces generating (i) axial and circumferential stress in the capillary walls that is balanced by the podocyte foot processes covering the capillary surface and by (ii) increased flow of the ultrafiltrate which, in turn, generates fluid flow shear stress (FFSS) on podocytes and their processes [105,106].
2.1. Fluid Flow Shear Stress (FFSS)
Fluid flow along the cell body and processes of podocytes is visualized as blood flow in the capillary and modeled using the flow of fluid over the flat surface of an object such as between two parallel plates or as fluid flow in a cylinder. Approximately 180 L/day of ultrafiltrate flows through human glomerular Bowman’s space that generates shear stress, causing cellular deformation of podocytes [105,106,107,108]. Fundamental considerations that form the basis of generating the plasma ultrafiltrate through the glomerular filtration barrier are based on hemodynamic parameters studied and established during the past several decades.
2.1.1. The Glomerular Filtration Barrier Function
Briefly, renal blood flow, ultrafiltration pressure (PUF), and ultrafiltration coefficient (Kf) are the main determinants of glomerular filtration. Kf (product of area and hydraulic permeability [Lp]) determines the Single Nephron Glomerular Filtration Rate (SNGFR) as shown in the equation.
In the above equation, x represents the normalized length of the glomerular capillary with the afferent-end designated by 0 and the efferent-end by 1; P and π represent the hydraulic and osmotic pressures in glomerular capillary (g) and Bowman space (B), respectively, at distance x along the capillary length; σ symbolizes the reflection coefficient (range 0 to 1). This equation shows that increase in Kf (determined by Lp and area) and/or ∆P (determined by PGC and ∆π) mainly determine the increase in single-nephron glomerular filtration rate (SNGFR) associated with glomerular hyperfiltration.
2.1.2. Unilateral Nephrectomy in Rodent Models of Hyperfiltration Increases Single-Nephron Glomerular Filtration Rate (SNGFR)
Hemodynamic parameters are utilized to study kidney disease in human subjects and animal models. Unilaterally nephrectomized (UNX) rodent models are valuable in determining hemodynamic changes and validating the calculated SNGFR in humans. Early studies showed that UNX in 5-day old rats (Sprague–Dawley) resulted in elevated SNGFR and PUF by 20 days of age, increased glomerular area and decreased Lp by 60 days of age. These findings also demonstrated that high ultrafiltration pressure was not an absolute requirement for increasing SNGFR in UNX rats. Additional studies showed that SNGFR increased by 30–36% in mice and 57–86% in rats [109,110,111]. Recent studies in our laboratory using mathematical modeling showed a 2-fold increase in SNGFR, a 2-fold increase in FFSS in UNX rats and a 2- to 3-fold increase in FFSS in UNX mice [73].
2.1.3. FFSS Mediates the Early Effects of Hyperfiltration
Shear stress due to ultrafiltrate flow targets the cell body and primary processes [107,112] and FFSS is considered the main cause of podocyte detachment and irreplaceable loss [112,113]. In this regard, FFSS-treated cultured podocytes show arrangement of the actin cytoskeleton with a cortical ring formation with increased PGE2 [114]. As mentioned, we demonstrated increased FFSS in UNX mice and rats [73].
2.2. Tensile Stress
The differential pressure (swelling pressure) between the capillary and Bowman space drives filtration while generating circumferential stress (~50 Kilopascals, kPa) and axial stress (0.3 kPa). Being much greater than axial stress, circumferential stress is considered the principal cause of tensile stress (calculated force of 2 nanonewtons) experienced in the podocyte foot process (0.04 µm2 area). The circumferential stress was shown to be borne by ≥20 actin filaments/foot process. Further, the capillary is assumed as a nonlinear elastic spring where the axial tension is countered by the elastic modulus of the structural elements of the capillary wall (collagen IV, laminin, etc.) with estimated Young’s modulus of 2000–5000 kPa [115].
Podocytes function as mechanosensors in response to mechanically induced stretch. The tensile stress due to capillary pressure causes podocytes to elongate along the GBM and develop a tensile strain to remain attached. Experimental biaxial stretch (0.5 Hz with 5% linear strain, 3 days) was shown to result in altered gene expression [105], activation of signaling cascades and generation/release of humoral factors and receptors [108,115,116,117].
Tensile Stress Alters the Actin Cytoskeleton Organization, Cell Adhesion, and Gene Expression of Podocytes
Capillary stretch targets slit junctions between foot processes [107,112]. Tensile stress has been shown to decrease transverse actin fibers and increase radial fibers in podocytes [115]. It also upregulates the secreted protein acidic and rich in cysteine (SPARC, i.e., Osteonectin) [118] and osteopontin [105,119] that attenuate the effect of cell stretch/detachment. In vitro studies showed that cell stretch induces p21, p38 MAPK, ERK1/2, and JNK [118,119,120,121,122,123,124,125]. We consider increased flow-induced shear as the primary of cellular stress in the early stages. However, increased tensile stress exacerbates the damage and accelerates the loss of function.
3. Tubulocentric and Podocentric Effects of Hyperfiltration
Deterioration of podocyte structure and glomerular function are primary and extensively studied features of hyperfiltration-induced changes during the early stages of glomerular disease. Physiological functions of glomerular and tubular segments are interdependent. Thus, both glomerular and tubular segments are subject to the effects of adaptive and maladaptive hyperfiltration and undergo changes that determine the pathogenesis of kidney disease.
3.1. Tubular Function and Glomerular Hyperfiltration
Tubular response to hyperfiltration and tubulo–glomerular interdependence are illustrated in several ways. Briefly, (i) tubular and glomerular components closely interact for the tubuloglomerular feedback (TGF) to regulate and maintain renal blood flow, GFR, and the tubular flow rate. [126]. (ii) Primary cilia in tubular cells function as mechano-sensors of hyperfiltration and activators of signaling [127]. (iii) Our collaborators have recently shown that hyperfiltration in unilaterally nephrectomized mice affects MAPK/ERK signaling, suggesting proliferation in principal cells of the collecting duct [128]. (iv) Glucose and sodium reabsorption by sodium-glucose co-transporters (SGLTs) in the proximal tubule increase due to tubular hyperplasia and hypertrophy, which, in turn, activates senescence-associated genes, inflammation and fibrosis [129]. (v) Sodium-glucose cotransporter-1 (SGLT1) activity in macula densa cells releases nitric oxide that promotes early hyperfiltration to recalibrate glucose and sodium levels [28]. (vi) SGLT2 inhibitors (SGLT2i) effectively normalize the tubuloglomerular feedback and lower hyperfiltration through inhibition of sodium and glucose reabsorption [130]. (vii) Persistent hyperfiltration increases tubular reabsorption and the cellular demand for oxygen that exacerbates oxidative stress [28,129]. (viii) The onset of albuminuria indicates either impaired reabsorption or ultrafiltrate protein levels exceeding the reabsorptive capacity for albumin by proximal tubular cells. Albuminuria exacerbates tubular stress [130]. (ix) Several molecules have been suggested as biomarkers of tubular stress and injury. These include N-acetyl-β-D-glucosaminidase, Neutrophil Gelatinase-Associated Lipocalin (NGAL) and Kidney Injury Molecule-1 (KIM1), urinary free Retinol-Binding Protein (RBP) and Cystatin C [131]. (x) Tubulointerstitial oxidative stress, inflammation, hypoxia and fibrosis relate to progression of kidney disease in diabetes [132].
3.2. Podocytes and Glomerular Hyperfiltration
Podocytes cover the GBM along the outer aspect of the capillary with a network of interdigitating foot processes in Bowman space. While the glomerular capillary is the site of hemodynamic changes, terminally differentiated and irreplaceable podocytes are immediately affected by hemodynamic changes. Podocytes provide the site of plasma filtration through slit pore junctions formed by their interdigitating foot processes, act as sensors of vascular changes and synthesize components of the glomerular basement membrane (GBM) that covers the capillary [133]. As discussed above, their location exposes podocytes to tensile stretch generated by capillary pressure and to shear stress due to ultrafiltrate flow along their apical, lateral, and basal surfaces [134].
We postulated that FFSS activates initial COX2-PGE2-EP2 signaling in podocytes. Overall, COX2-PGE2-EP2 axis with the activation of AKT-GSK3β-β catenin and c-Src-PLD-mTOR signaling mediates the early effects of FFSS [134]. We also demonstrated the ex vivo effect of FFSS on podocytes in intact glomeruli where FFSS or PGE2 caused an increase in glomerular albumin permeability of isolated rat glomeruli that was blocked by indomethacin, an inhibitor of prostaglandin synthesis [73]. Additional work using unilaterally nephrectomized sv129 mice that have normal nephron endowment and Os/+ mice that are born with low nephron number showed increased albuminuria and glomerular expression of COX2 and PGE2 receptor EP2 proteins [73,135].
Glomerular Hyperfiltration Results in Podocytes Loss
Overall, hyperfiltration-associated stress induces glomerular hypertrophy. However, podocyte hypertrophy is not in proportion with the increase in the GBM length. Secondly, podocytes respond to stress (e.g., hyperfiltration) by contracting actin fibers, resulting in denudation of the GBM [136]. These disparate changes in GBM and podocytes result in: (i) areas of the GBM left uncovered by podocyte processes adding to podocyte injury, detachment and loss; (ii) adherence of the capillary to parietal epithelium; (iii) formation of synechia and segmental sclerosis [27,113]. Viable podocytes were detected in urine samples following detachment from GBM in patients with FSGS [137], IgA nephropathy [138] and diabetes [139]. Detachment and loss of podocytes exacerbates proteinuria in these conditions.
Cellular adhesion to the extracellular matrix involves membrane glycoproteins and proteoglycans [140]. Podocytes cover the outer aspect of the matrix through interdigitating foot process with slit pore junctions and also contribute to matrix structure by synthesizing its components. We previously showed that FFSS caused derangement of the actin cytoskeleton in podocytes, activated the COX2-PGE2-EP2 signaling axis and resulted in Akt-glycogen synthase kinase-3β-β-catenin and MAPK/ERK signaling. We recently used IMPres analysis, a novel bioinformatics algorithm for hypotheses generation using pathway networks and multi-omics data [141]. This analysis showed that activation of the COX2, EP2, and β-catenin is linked to changes in proteoglycans and galactose metabolism in FFSS-treated podocytes involved in glycocalyx remodeling and cell attachment/detachment [142].
Thus, increased fluid flow during early hyperfiltration affects the size, shape and adhesion of podocytes. Ultrastructural integrity of podocytes is essential for the glomerular filtration barrier function. As mentioned, we have shown that COX2-PGE2-EP2 axis, β-catenin signaling and glycocalyx remodeling pathways are upregulated in podocytes exposed to fluid flow shear stress. Therefore, we are pursuing the idea that biomechanical stress triggers signaling events that begin at the plasma membrane.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10020407
This entry is offline, you can click here to edit this entry!