Genetic predisposition, as in other inflammatory diseases, might be responsible for alterations in the clinical course of COVID-19 patients through polymorphisms in crucial genes such as ACE2 and MHC class I. Components of the immune response to the virus appear to be primarily related to disease severity, whereas genes related to the binding of the ACE2 cell surface—the entry point for SARS-CoV-2—during the early stages of infection appear to be largely responsible for the varying susceptibility to SARS-CoV-2. Inflammatory inhibitors are at the forefront of pharmacological management in COVID-19, although their potential has not been fully elucidated till now. The above mentioned would have a potentially large impact on targeted medicines and, more critically, vaccine development.
- COVID-19
- SARS-CoV-2
- inflammation
- genetics
- cytokines
1. Introduction
2. Cytokine Storm and ARDS in COVID-19
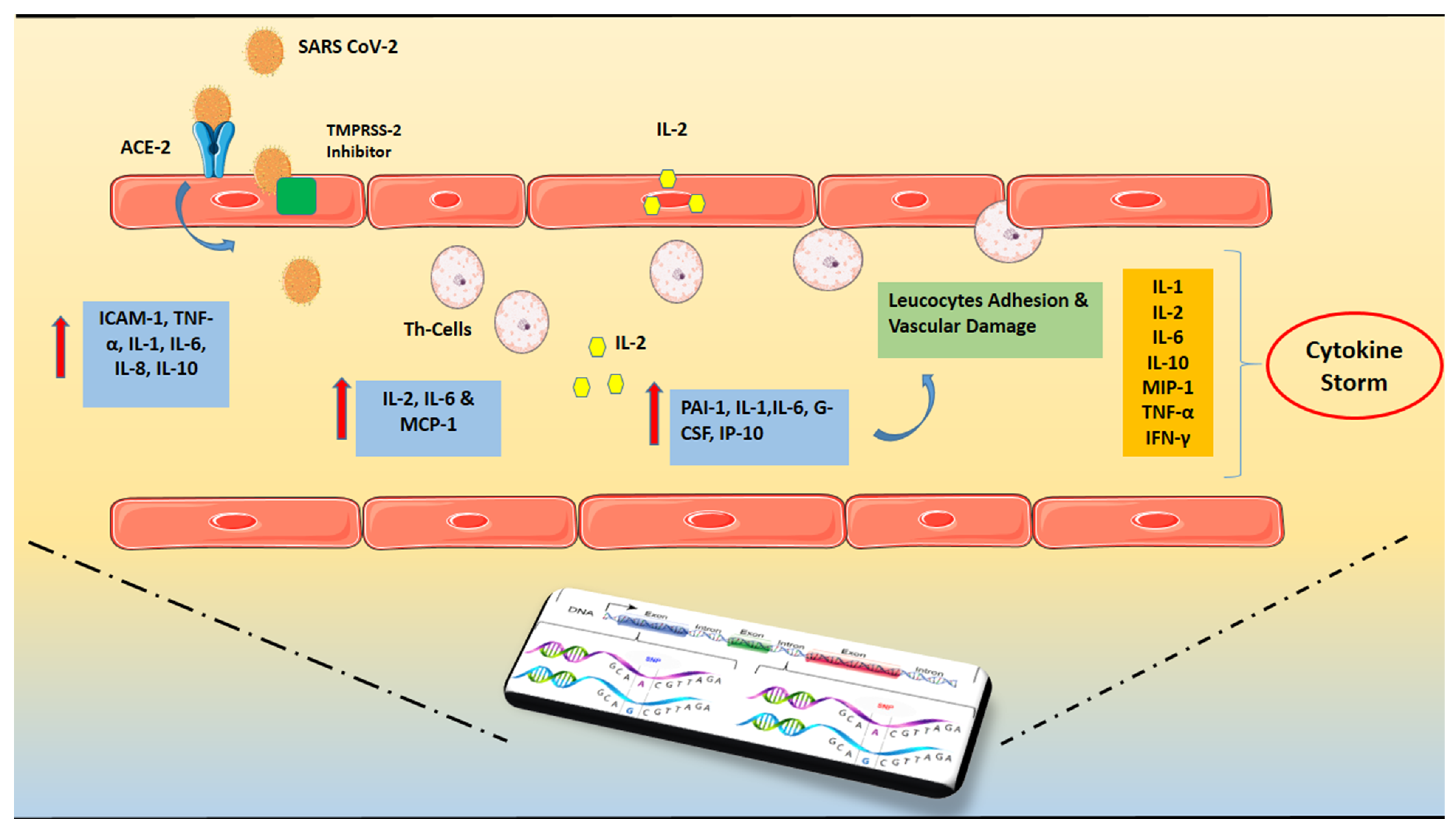 Figure 1. SARS-CoV-2 invasion and hyper-inflammatory state in close relation with genetic predisposition. The presence of Angiotensin-converting enzyme 2 (ACE2) and Transmembrane protease serine 2 (TMPRSS-2) that may cleave the viral spike is required for SARS-CoV-2’s cell invasion. Increased levels of pro-inflammatory cytokines, particularly the soluble interleukin 2-receptor (IL-2R) and interleukin-6 (IL-6) have been found. Soluble IL-2R (sIL-2R) is mostly released by activated T helper lymphocytes, although it may also be secreted by endothelial cells (ECs). The capillary leak is caused by the binding of IL-6 and IL-2 to their receptors. The persistent burdening of the endothelium results in increased release of inflammatory cytokines and immune system overreaction, resulting in the so-called “cytokine storm”. The above mentioned hyper-inflammatory state is in close relation with the individual genetic profile which can potentially govern the course of the disease. Abbreviations: SARS CoV-2 = Severe acute respiratory syndrome Coronavirus-2, ACE2 = Angiotensin-converting enzyme 2, TMPRSS = Transmembrane protease serine 2, IL = Interleukin, ΡAΙ-1 = Plasminogen activator inhibitor-1, TNF = Tumor Necrosis Factor, ICAM = Intercellular Adhesion Molecule 1, MCP-1 = monocyte chemoattractant protein-1, G-CSF = Granulocyte colony-stimulating factor, IP-10 = Interferon gamma-induced protein 10, MIP-1 = Macrophage inflammatory protein-1, IFN = Interferon.
Figure 1. SARS-CoV-2 invasion and hyper-inflammatory state in close relation with genetic predisposition. The presence of Angiotensin-converting enzyme 2 (ACE2) and Transmembrane protease serine 2 (TMPRSS-2) that may cleave the viral spike is required for SARS-CoV-2’s cell invasion. Increased levels of pro-inflammatory cytokines, particularly the soluble interleukin 2-receptor (IL-2R) and interleukin-6 (IL-6) have been found. Soluble IL-2R (sIL-2R) is mostly released by activated T helper lymphocytes, although it may also be secreted by endothelial cells (ECs). The capillary leak is caused by the binding of IL-6 and IL-2 to their receptors. The persistent burdening of the endothelium results in increased release of inflammatory cytokines and immune system overreaction, resulting in the so-called “cytokine storm”. The above mentioned hyper-inflammatory state is in close relation with the individual genetic profile which can potentially govern the course of the disease. Abbreviations: SARS CoV-2 = Severe acute respiratory syndrome Coronavirus-2, ACE2 = Angiotensin-converting enzyme 2, TMPRSS = Transmembrane protease serine 2, IL = Interleukin, ΡAΙ-1 = Plasminogen activator inhibitor-1, TNF = Tumor Necrosis Factor, ICAM = Intercellular Adhesion Molecule 1, MCP-1 = monocyte chemoattractant protein-1, G-CSF = Granulocyte colony-stimulating factor, IP-10 = Interferon gamma-induced protein 10, MIP-1 = Macrophage inflammatory protein-1, IFN = Interferon.3. Genetic Predisposition
| Gene | Polymorphism | Result |
|---|---|---|
| ABO | rs657152 | Higher risk of infection for blood group A vs. non-A and lower risk of infection for blood group O vs. non-O [26]. |
| HLA | HLA-B*46:01, HLA-A*11:01, -B*51:01, -C*14:02, HLA-DRB1*15:01, -DQB1*06:02, and -B*27:07 | Vulnerable to disease for HLA-B*46:01 and cross-protective T cell-based immunity for HLA-B*15:03 [39]. |
| TMPRSS2 | p.Val160Met (rs12329760) | Increased susceptibility to SARS-CoV-2 [35]. |
| ACE2 | K31R, N33I, H34R, E35K, E37K, D38V, Y50F, N51S, M62V, K68E, F72V, Y83H, G326E, G352V, D355N, Q388L, and D509Y rs233574 rs2074192 rs4646188 (p.(Asn720Asp) p.(Lys26Arg) p.(Gly211Arg) p.(Leu351Val) p.(Pro389His) |
Better cardiovascular and pulmonary course of the disease, less susceptibility to SARS-CoV-2 [31][32][33][34]. |
| ApoE | rs429358-C-C (e4e4) | Severe course of the disease [37][38]. |
| SLC6A20, LZTFL1, CCR9, FYCO1, CXCR6, XCR1 | rs11385942-GA | Severe course of the disease and potentially higher odds for ARDS [42][43]. |
| Of Complement proteins | C3 FF, C3 FS, C3 SS | Severe course of the disease, Increased susceptibility to SARS-CoV-2 [41]. |
| TMEM189- UBE2V1 | rs6020298-A | Severe course of the disease [40]. |
| TLR7 | g.12905756_12905759del and g.12906010G > T | Severe course of the disease [39]. |
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10020242
References
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-up. J. Am. Coll. Cardiol. 2020, 75, 2950–2973.
- Harapan, H.; Itoh, N.; Yufika, A.; Winardi, W.; Keam, S.; Te, H.; Megawati, D.; Hayati, Z.; Wagner, A.L.; Mudatsir, M. Coronavirus disease 2019 (COVID-19): A literature review. J. Infect. Public Health 2020, 13, 667–673.
- The WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group; Sterne, J.A.C.; Murthy, S.; Diaz, J.V.; Slutsky, A.S.; Villar, J.; Angus, D.C.; Annane, D.; Azevedo, L.C.P.; Berwanger, O.; et al. Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-analysis. JAMA 2020, 324, 1330–1341.
- Abbasi-Oshaghi, E.; Mirzaei, F.; Farahani, F.; Khodadadi, I.; Tayebinia, H. Diagnosis and treatment of coronavirus disease 2019 (COVID-19): Laboratory, PCR, and chest CT imaging findings. Int. J. Surg. 2020, 79, 143–153.
- Siddiqi, H.K.; Mehra, M.R. COVID-19 illness in native and immunosuppressed states: A clinical-therapeutic staging proposal. J. Heart Lung Transpl. 2020, 39, 405–407.
- Sagris, M.; Theofilis, P.; Antonopoulos, A.S.; Tsioufis, C.; Oikonomou, E.; Antoniades, C.; Crea, F.; Kaski, J.C.; Tousoulis, D. Inflammatory Mechanisms in COVID-19 and Atherosclerosis: Current Pharmaceutical Perspectives. Int. J. Mol. Sci. 2021, 22, 6607.
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613.
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034.
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518.
- Machowicz, R.; Janka, G.; Wiktor-Jedrzejczak, W. Similar but not the same: Differential diagnosis of HLH and sepsis. Crit. Rev. Oncol. Hematol. 2017, 114, 1–12.
- Amaral, M.C.; Alves, J.D. Pathogenesis of multi-organic failure in autoimmune diseases. Autoimmun. Rev. 2009, 8, 525–528.
- Theofilis, P.; Sagris, M.; Antonopoulos, A.S.; Oikonomou, E.; Tsioufis, C.; Tousoulis, D. Inflammatory Mediators of Platelet Activation: Focus on Atherosclerosis and COVID-19. Int. J. Mol. Sci. 2021, 22, 11170.
- Alunno, A.; Carubbi, F.; Rodríguez-Carrio, J. Storm, typhoon, cyclone or hurricane in patients with COVID-19? Beware of the same storm that has a different origin. RMD Open 2020, 6, e001295.
- Mazodier, K.; Marin, V.; Novick, D.; Farnarier, C.; Robitail, S.; Schleinitz, N.; Veit, V.; Paul, P.; Rubinstein, M.; Dinarello, C.A.; et al. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood 2005, 106, 3483–3489.
- Cheung, C.Y.; Poon, L.L.; Ng, I.H.; Luk, W.; Sia, S.F.; Wu, M.H.; Chan, K.H.; Yuen, K.Y.; Gordon, S.; Guan, Y.; et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: Possible relevance to pathogenesis. J. Virol. 2005, 79, 7819–7826.
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Frydas, I.; Kritas, S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34, 1.
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781.
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848.
- Lau, S.K.P.; Lau, C.C.Y.; Chan, K.H.; Li, C.P.Y.; Chen, H.; Jin, D.Y.; Chan, J.F.W.; Woo, P.C.Y.; Yuen, K.Y. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: Implications for pathogenesis and treatment. J. Gen. Virol. 2013, 94, 2679–2690.
- Law, H.K.; Cheung, C.Y.; Ng, H.Y.; Sia, S.F.; Chan, Y.O.; Luk, W.; Nicholls, J.M.; Peiris, J.S.; Lau, Y.L. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood 2005, 106, 2366–2374.
- Li, G.; Hu, R.; Gu, X. A close-up on COVID-19 and cardiovascular diseases. Nutr. Metab. Cardiovasc. Dis. NMCD 2020, 30, 1057–1060.
- Hogner, K.; Wolff, T.; Pleschka, S.; Plog, S.; Gruber, A.D.; Kalinke, U.; Walmrath, H.D.; Bodner, J.; Gattenlohner, S.; Lewe-Schlosser, P.; et al. Correction: Macrophage-expressed IFN-beta Contributes to Apoptotic Alveolar Epithelial Cell Injury in Severe Influenza Virus Pneumonia. PLoS Pathog. 2016, 12, e1005716.
- Rodrigue-Gervais, I.G.; Labbe, K.; Dagenais, M.; Dupaul-Chicoine, J.; Champagne, C.; Morizot, A.; Skeldon, A.; Brincks, E.L.; Vidal, S.M.; Griffith, T.S.; et al. Cellular inhibitor of apoptosis protein cIAP2 protects against pulmonary tissue necrosis during influenza virus infection to promote host survival. Cell Host. Microbe 2014, 15, 23–35.
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537.
- Sagris, M.; Vardas, E.P.; Theofilis, P.; Antonopoulos, A.S.; Oikonomou, E.; Tousoulis, D. Atrial Fibrillation: Pathogenesis, Predisposing Factors, and Genetics. Int. J. Mol. Sci. 2021, 23, 6.
- Severe Covid, G.G.; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernandez, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe COVID-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534.
- Wu, Y.; Feng, Z.; Li, P.; Yu, Q. Relationship between ABO blood group distribution and clinical characteristics in patients with COVID-19. Clin. Chim. Acta 2020, 509, 220–223.
- Zhao, J.; Yang, Y.; Huang, H.; Li, D.; Gu, D.; Lu, X.; Zhang, Z.; Liu, L.; Liu, T.; Liu, Y.; et al. Relationship between the ABO Blood Group and the Coronavirus Disease 2019 (COVID-19) Susceptibility. Clin. Infect. Dis. 2021, 73, 328–331.
- Diavati, S.; Sagris, M.; Terentes-Printzios, D.; Vlachopoulos, C. Anticoagulation Treatment in Venous Thromboembolism: Options and Optimal Duration. Curr. Pharm. Des. 2021.
- Wu, B.B.; Gu, D.Z.; Yu, J.N.; Yang, J.; Shen, W.Q. Association between ABO blood groups and COVID-19 infection, severity and demise: A systematic review and meta-analysis. Infect. Genet. Evol. 2020, 84, 104485.
- Hou, Y.; Zhao, J.; Martin, W.; Kallianpur, A.; Chung, M.K.; Jehi, L.; Sharifi, N.; Erzurum, S.; Eng, C.; Cheng, F. New insights into genetic susceptibility of COVID-19: An ACE2 and TMPRSS2 polymorphism analysis. BMC Med. 2020, 18, 216.
- Suryamohan, K.; Diwanji, D.; Stawiski, E.W.; Gupta, R.; Miersch, S.; Liu, J.; Chen, C.; Jiang, Y.P.; Fellouse, F.A.; Sathirapongsasuti, J.F.; et al. Human ACE2 receptor polymorphisms and altered susceptibility to SARS-CoV-2. Commun. Biol. 2021, 4, 475.
- Pouladi, N.; Abdolahi, S. Investigating the ACE2 polymorphisms in COVID-19 susceptibility: An in silico analysis. Mol. Genet. Genom. Med. 2021, 9, e1672.
- Novelli, A.; Biancolella, M.; Borgiani, P.; Cocciadiferro, D.; Colona, V.L.; D’Apice, M.R.; Rogliani, P.; Zaffina, S.; Leonardis, F.; Campana, A.; et al. Analysis of ACE2 genetic variants in 131 Italian SARS-CoV-2-positive patients. Hum. Genom. 2020, 14, 29.
- Vargas-Alarcon, G.; Posadas-Sanchez, R.; Ramirez-Bello, J. Variability in genes related to SARS-CoV-2 entry into host cells (ACE2, TMPRSS2, TMPRSS11A, ELANE, and CTSL) and its potential use in association studies. Life Sci. 2020, 260, 118313.
- Siasos, G.; Skotsimara, G.; Oikonomou, E.; Sagris, M.; Vasiliki-Chara, M.; Bletsa, E.; Stampouloglou, P.; Theofilis, P.; Charalampous, G.; Tousoulis, D. Antithrombotic Treatment in Diabetes Mellitus: A Review of the Literature about Antiplatelet and Anticoagulation Strategies Used for Diabetic Patients in Primary and Secondary Prevention. Curr. Pharm. Des. 2020, 26, 2780–2788.
- Kasparian, K.; Graykowski, D.; Cudaback, E. Commentary: APOE e4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. Front. Immunol. 2020, 11, 1939.
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Kuchel, G.A.; Melzer, D. APOE e4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2231–2232.
- Nguyen, A.; David, J.K.; Maden, S.K.; Wood, M.A.; Weeder, B.R.; Nellore, A.; Thompson, R.F. Human Leukocyte Antigen Susceptibility Map for Severe Acute Respiratory Syndrome Coronavirus 2. J. Virol. 2020, 94, e00510-20.
- Wang, F.; Huang, S.; Gao, R.; Zhou, Y.; Lai, C.; Li, Z.; Xian, W.; Qian, X.; Li, Z.; Huang, Y.; et al. Initial whole-genome sequencing and analysis of the host genetic contribution to COVID-19 severity and susceptibility. Cell Discov. 2020, 6, 83.
- Delanghe, J.R.; De Buyzere, M.L.; Speeckaert, M.M. Genetic Polymorphisms in the Host and COVID-19 Infection. Adv. Exp. Med. Biol. 2021, 1318, 109–118.
- Thevarajan, I.; Nguyen, T.H.O.; Koutsakos, M.; Druce, J.; Caly, L.; van de Sandt, C.E.; Jia, X.; Nicholson, S.; Catton, M.; Cowie, B.; et al. Breadth of concomitant immune responses prior to patient recovery: A case report of non-severe COVID-19. Nat. Med. 2020, 26, 453–455.
- van der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; van den Heuvel, G.; Mantere, T.; Kersten, S.; van Deuren, R.C.; Steehouwer, M.; van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of Genetic Variants Among Young Men with Severe COVID-19. JAMA 2020, 324, 663–673.