Extracellular vesicles (EVs) are defined as a heterogenic group of lipid bilayer vesicular structures with a size in the range of 30–4000 nm that are released by all types of cultured cells. EVs derived from platelets, mononuclears, endothelial cells, and adipose tissue cells significantly increase in several cardiovascular diseases, including in atrial fibrillation (AF). EVs are engaged in cell-to-cell cooperation, endothelium integrity, inflammation, and immune response and are a cargo for several active molecules, such as regulatory peptides, receptors, growth factors, hormones, and lipids. Being transductors of the intercellular communication, EVs regulate angiogenesis, neovascularization, coagulation, and maintain tissue reparation.
- extracellular vesicles
- atrial fibrillation
- thrombogenicity
- biomarkers
- prediction
1. Introduction
2. Extracellular Vesicles: Definition, Nomenclature, Biological Function
| Characteristics | Exosomes | Microvesicles | Apoptotic Bodies |
|---|---|---|---|
| Diameter, nm | 30–150 | 100–1000 | 500–4000 |
| Sedimentation, g | 100,000 | 20,000 | 16,000 |
| Pathway for biogenesis | Endocytosis from endosomes and exocytosis of late endosomes/MVBs | Blebbing from plasma membranes | Shrinkage and blebbing of apoptotic cells |
| Unconventional secretion pathway | Cellular activation | Early apoptosis | Lysosome vesicle secretion and secretory autophagy |
| Delivery contents | Alix, chaperones, Rab proteins, Rab GTPases, SNAREs, lipid rafts, proteins (flotillin), myokines, inflammatory cytokines, growth factors, miRs. | Arachidonic acid, cytokines, chemokine RANTES/CCL5, P-selectin, lipids, signaling proteins, miRNA, and microRNA, membrane-anchored receptors (PPARγ) and adhesion molecules | Organelles and/or nuclear content including chromatin, DNA, miRNAs, microRNAs, histones, oncogenes. |
| Membrane-specific antigens | Tetraspanins (CD9, CD81, CD63), TSG101, | Integrins, selectins, membrane proteins of parental cells | Annexin-V(+) |
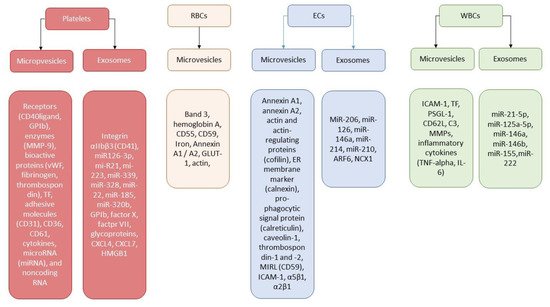
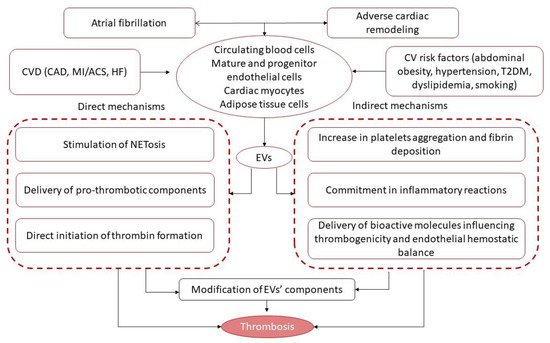
3. Extracellular Vesicles and Thrombogenicity/Thrombosis
This entry is adapted from the peer-reviewed paper 10.3390/ijms23031774
References
- Di Carlo, A.; Bellino, L.; Consoli, D.; Mori, F.; Zaninelli, A.; Baldereschi, M.; Cattarinussi, A.; D’Alfonso, M.G.; Gradia, C.; Sgherzi, B.; et al. Prevalence of atrial fibrillation in the Italian elderly population and projections from 2020 to 2060 for Italy and the European Union: The FAI Project. EP Eur. 2019, 21, 1468–1475.
- Tedrow, U.B.; Conen, D.; Ridker, P.M.; Cook, N.R.; Koplan, B.A.; Manson, J.E.; Buring, J.E.; Albert, C.M. The long- and short-term impact of elevated body mass index on the risk of new atrial fibrillation the WHS (women’s health study). J. Am. Coll. Cardiol. 2010, 55, 2319–2327.
- Kloosterman, M.; Crijns, H.; Van Gelder, I.C. Rising prevalence of atrial fibrillation in the elderly population: New challenges of geriatric cardiology. EP Eur. 2019, 21, 1451–1453.
- Carlisle, M.A.; Fudim, M.; DeVore, A.D.; Piccini, J.P. Heart Failure and Atrial Fibrillation, Like Fire and Fury. JACC Heart Fail. 2019, 7, 447–456.
- Hua, C.; Jiang, C.; He, L.; Jia, Z.X.; Lyu, W.H.; Tang, R.B.; Sang, C.H.; Long, D.Y.; Dong, J.Z.; Ma, C.S.; et al. Causes of death and influencing factors of atrial fibrillation patients undergoing anticoagulation therapy. Zhonghua Xin Xue Guan Bing Za Zhi 2021, 49, 353–359.
- Rankin, A.J.; Rankin, S.H. Cardioverting acute atrial fibrillation and the risk of thromboembolism: Not all patients are created equal. Clin. Med. 2017, 17, 419–423.
- Proietti, M.; Laroche, C.; Drozd, M.; Vijgen, J.; Cozma, D.C.; Drozdz, J.; Maggioni, A.P.; Boriani, G.; Lip, G.Y.H.; EORP-AF Investigators. Impact of chronic obstructive pulmonary disease on prognosis in atrial fibrillation: A report from the EURObservational Research Programme Pilot Survey on Atrial Fibrillation (EORP-AF) General Registry. Am. Heart J. 2016, 181, 83–91.
- Boriani, G.; Laroche, C.; Diemberger, I.; Fantecchi, E.; Popescu, M.I.; Rasmussen, L.H.; Dan, G.-A.; Kalarus, Z.; Tavazzi, L.; Maggioni, A.P.; et al. ‘Real-world’ management and outcomes of patients with paroxysmal vs. non-paroxysmal atrial fibrillation in Europe: The EURObservational Research Programme-Atrial Fibrillation (EORP-AF) General Pilot Registry. EP Eur. 2016, 18, 648–657.
- Lip, G.Y.H.; Laroche, C.; Boriani, G.; Cimaglia, P.; Dan, G.-A.; Santini, M.; Kalarus, Z.; Rasmussen, L.H.; Popescu, M.I.; Tica, O.; et al. Sex-related differences in presentation, treatment, and outcome of patients with atrial fibrillation in Europe: A report from the Euro Observational Research Programme Pilot survey on Atrial Fibrillation. EP Eur. 2015, 17, 24–31.
- Sivasambu, B.; Balouch, M.A.; Zghaib, T.; Bajwa, R.J.; Chrispin, J.; Berger, R.D.; Ashikaga, H.; Nazarian, S.; Marine, J.E.; Calkins, H.; et al. Increased rates of atrial fibrillation recurrence following pulmonary vein isolation in overweight and obese patients. J. Cardiovasc. Electrophysiol. 2018, 29, 239–245.
- Scherr, D.; Khairy, P.; Miyazaki, S.; Aurillac-Lavignolle, V.; Pascale, P.; Wilton, S.B.; Ramoul, K.; Komatsu, Y.; Roten, L.; Jadidi, A.; et al. Five-year outcome of catheter ablation of persistent atrial fibrillation using termination of atrial fibrillation as a procedural endpoint. Circulation. Arrhythmia Electrophysiol. 2015, 8, 18–24.
- Saguner, A.M.; Maurer, T.; Wissner, E.; Santoro, F.; Lemes, C.; Mathew, S.; Sohns, C.; Heeger, C.H.; Reißmann, B.; Riedl, J.; et al. Catheter ablation of atrial fibrillation in very young adults: A 5-year follow-up study. EP Eur. 2018, 20, 58–64.
- Jame, S.; Barnes, G. Stroke and thromboembolism prevention in atrial fibrillation. Heart 2020, 106, 10–17.
- O’Neill, L.; Wielandts, J.Y.; Gillis, K.; Hilfiker, G.; Le Polain De Waroux, J.-B.; Tavernier, R.; Duytschaever, M.; Knecht, S. Catheter Ablation in Persistent AF, the Evolution towards a More Pragmatic Strategy. J. Clin. Med. 2021, 10, 4060.
- Blanch-Ruiz, M.A.; Ortega-Luna, R.; Martínez-Cuesta, M.Á.; Álvarez, Á. The Neutrophil Secretome as a Crucial Link between Inflammation and Thrombosis. Int. J. Mol. Sci. 2021, 22, 4170.
- Berezin, A.; Zulli, A.; Kerrigan, S.; Petrovic, D.; Kruzliak, P. Predictive role of circulating endothelial-derived microparticles in cardiovascular diseases. Clin. Biochem. 2015, 48, 562–568.
- De Freitas, R.; Hirata, R.; Hirata, M.H.; Aikawa, E. Circulating Extracellular Vesicles as Biomarkers and Drug Delivery Vehicles in Cardiovascular Diseases. Biomolecules 2021, 11, 388.
- Théry, C.; Witwer, K.W. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750.
- Van der Pol, E.; Böing, A.N.; Gool, E.L.; Nieuwland, R. Recent developments in the nomenclature, presence, isolation, detection and clinical impact of extracellular vesicles. J. Thromb. Haemost. JTH 2016, 14, 48–56.
- Nederveen, J.P.; Warnier, G.; Di Carlo, A.; Nilsson, M.I.; Tarnopolsky, M.A. Extracellular Vesicles and Exosomes: Insights from Exercise Science. Front. Physiol. 2021, 11, 604274.
- Askeland, A.; Borup, A.; Østergaard, O.; Olsen, J.V.; Lund, S.M.; Christiansen, G.; Kristensen, S.R.; Heegaard, N.; Pedersen, S. Mass-Spectrometry Based Proteome Comparison of Extracellular Vesicle Isolation Methods: Comparison of ME-kit, Size-Exclusion Chromatography, and High-Speed Centrifugation. Biomedicines 2020, 8, 246.
- Witwer, K.W.; Goberdhan, D.C.; O’Driscoll, L.; Théry, C.; Welsh, J.A.; Blenkiron, C.; Buzás, E.I.; Di Vizio, D.; Erdbrügger, U.; Falcón-Pérez, J.M.; et al. Updating MISEV: Evolving the minimal requirements for studies of extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12182.
- Femminò, S.; Penna, C.; Margarita, S.; Comità, S.; Brizzi, M.F.; Pagliaro, P. Extracellular vesicles and cardiovascular system: Biomarkers and Cardioprotective Effectors. Vasc. Pharmacol. 2020, 135, 106790.
- Puhm, F.; Boilard, E.; Machlus, K.R. Platelet Extracellular Vesicles: Beyond the Blood. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 87–96.
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312.
- Puhm, F.; Flamand, L.; Boilard, E. Platelet extracellular vesicles in COVID-19: Potential markers and makers. J. Leukoc. Biol. 2022, 111, 63–74.
- Berezin, A.E. Microparticles in Chronic Heart Failure. Adv. Clin. Chem. 2017, 81, 1–41.
- Bebelman, M.P.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11.
- Arraud, N.; Linares, R.; Tan, S.; Gounou, C.; Pasquet, J.M.; Mornet, S.; Brisson, A.R. Extracellular vesicles from blood plasma: Determination of their morphology, size, phenotype and concentration. J. Thromb. Haemost. 2014, 12, 614–627.
- Sinauridze, E.I.; Kireev, D.A.; Popenko, N.Y.; Pichugin, A.V.; Panteleev, M.A.; Krymskaya, O.V.; Ataullakhanov, F.I. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb. Haemost. 2007, 97, 425–434.
- Simons, M.; Raposo, G. Exosomes—Vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581.
- Laffont, B.; Corduan, A.; Rousseau, M.; Duchez, A.C.; Lee, C.H.; Boilard, E.; Provost, P. Platelet microparticles reprogram macrophage gene expression and function. Thromb. Haemost. 2016, 115, 311–323.
- Silverman-Gavrila, R.; Silverman-Gavrila, L.; Bendeck, M.P. Cell division fidelity is altered during the vascular response to injury: Its novel role in atherosclerosis progression. Am. J. Pathol. 2013, 182, 628–639.
- Fourcade, O.; Simon, M.F.; Viode, C.; Rugani, N.; Leballe, F.; Ragab, A.; Fournie, B.; Sarda, L.; Chap, H. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell 1995, 80, 919–927.
- Almeida, V.H.; Rondon, A.; Gomes, T.; Monteiro, R.Q. Novel Aspects of Extracellular Vesicles as Mediators of Cancer-Associated Thrombosis. Cells 2019, 8, 716.
- Berezin, A. Neutrophil extracellular traps: The core player in vascular complications of diabetes mellitus. Diabetes Metab. Syndr. 2019, 13, 3017–3023.
- Gasecka, A.; Böing, A.N.; Filipiak, K.J.; Nieuwland, R. Platelet extracellular vesicles as biomarkers for arterial thrombosis. Platelets 2017, 28, 228–234.
- Rothmeier, A.S.; Versteeg, H.H.; Ruf, W. Factor VIIa-induced interaction with integrin controls the release of tissue factor on extracellular vesicles from endothelial cells. J. Thromb. Haemost. 2019, 17, 627–634.
- Rothmeier, A.S.; Liu, E.; Chakrabarty, S.; Disse, J.; Mueller, B.M.; Østergaard, H.; Ruf, W. Identification of the integrin-binding site on coagulation factor VIIa required for proangiogenic PAR2 signaling. Blood 2018, 131, 674–685.
- Zifkos, K.; Dubois, C.; Schäfer, K. Extracellular Vesicles and Thrombosis: Update on the Clinical and Experimental Evidence. Int. J. Mol. Sci. 2021, 22, 9317.
- Rosas, M.; Slatter, D.A.; Obaji, S.G.; Webber, J.P.; Alvarez-Jarreta, J.; Thomas, C.P.; Aldrovandi, M.; Tyrrell, V.J.; Jenkins, P.V.; O’Donnell, V.B.; et al. The procoagulant activity of tissue factor expressed on fibroblasts is increased by tissue factor-negative extracellular vesicles. PLoS ONE 2020, 15, e0240189.
- Gąsecka, A.; Rogula, S.; Eyileten, C.; Postuła, M.; Jaguszewski, M.J.; Kochman, J.; Mazurek, T.; Nieuwland, R.; Filipiak, K.J. Role of P2Y Receptors in Platelet Extracellular Vesicle Release. Int. J. Mol. Sci. 2020, 21, 6065.
- Zarà, M.; Guidetti, G.F.; Camera, M.; Canobbio, I.; Amadio, P.; Torti, M.; Tremoli, E.; Barbieri, S.S. Biology and Role of Extracellular Vesicles (EVs) in the Pathogenesis of Thrombosis. Int. J. Mol. Sci. 2019, 20, 2840.
- Suades, R.; Padro, T.; Vilahur, G.; Badimon, L. Circulating and platelet-derived microparticles in human blood enhance thrombosis on atherosclerotic plaques. Thromb. Haemost. 2012, 108, 1208–1219.
- Barry, O.P.; Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J. Clin. Investig. 1997, 99, 2118–2127.
- Caughey, G.E.; Cleland, L.G.; Gamble, J.R.; James, M.J. Up-regulation of endothelial cyclooxygenase-2 and prostanoid synthesis by platelets. Role of thromboxane A2. J. Biol. Chem. 2001, 276, 37839–37845.
- Aharon, A.; Tamari, T.; Brenner, B. Monocyte-derived microparticles and exosomes induce procoagulant and apoptotic effects on endothelial cells. Thromb. Haemost. 2008, 100, 878–885.
- Barry, O.P.; Kazanietz, M.G.; Praticò, D.; FitzGerald, G.A. Arachidonic acid in platelet microparticles up-regulates cyclooxygenase-2-dependent prostaglandin formation via a protein kinase C/mitogen-activated protein kinase-dependent pathway. J. Biol. Chem. 1999, 274, 7545–7556.
- Lacroix, R.; Sabatier, F.; Mialhe, A.; Basire, A.; Pannell, R.; Borghi, H.; Robert, S.; Lamy, E.; Plawinski, L.; Camoin-Jau, L.; et al. Activation of plasminogen into plasmin at the surface of endothelial microparticles: A mechanism that modulates angiogenic properties of endothelial progenitor cells in vitro. Blood 2007, 110, 2432–2439.
- Plesner, T.; Behrendt, N.; Ploug, M. Structure, function and expression on blood and bone marrow cells of the urokinase-type plasminogen activator receptor, uPAR. Stem cells 1997, 15, 398–408.
- Kwon, M.; MacLeod, T.J.; Zhang, Y.; Waisman, D.M. S100A10, annexin A2, and annexin a2 heterotetramer as candidate plasminogen receptors. Front. Biosci. 2005, 10, 300–325.
- He, K.L.; Deora, A.B.; Xiong, H.; Ling, Q.; Weksler, B.B.; Niesvizky, R.; Hajjar, K.A. Endothelial cell annexin A2 regulates polyubiquitination and degradation of its binding partner S100A10/p11. J. Biol. Chem. 2008, 283, 19192–19200.
- Miller, V.A.; Madureira, P.A.; Kamaludin, A.A.; Komar, J.; Sharma, V.; Sahni, G.; Thelwell, C.; Longstaff, C.; Waisman, D.M. Mechanism of plasmin generation by S100A10. Thromb. Haemost. 2017, 117, 1058–1071.
- Bharadwaj, A.; Kempster, E.; Waisman, D.M. The Annexin A2/S100A10 Complex: The Mutualistic Symbiosis of Two Distinct Proteins. Biomolecules 2021, 11, 1849.
- Stojkovic, S.; Thulin, Å.; Hell, L.; Thaler, B.; Rauscher, S.; Baumgartner, J.; Gröger, M.; Ay, C.; Demyanets, S.; Neumayer, C.; et al. IL-33 stimulates the release of procoagulant microvesicles from human monocytes and differentially increases tissue factor in human monocyte subsets. Thromb. Haemost. 2017, 117, 1379–1390.
- Liu, M.L.; Reilly, M.P.; Casasanto, P.; McKenzie, S.E.; Williams, K.J. Cholesterol enrichment of human monocyte/macrophages induces surface exposure of phosphatidylserine and the release of biologically-active tissue factor-positive microvesicles. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 430–435.
- Harvey, K.; Siddiqui, R.A.; Sliva, D.; Garcia, J.G.; English, D. Serum factors involved in human microvascular endothelial cell morphogenesis. J. Lab. Clin. Med. 2002, 140, 188–198.
- English, D.; Welch, Z.; Kovala, A.T.; Harvey, K.; Volpert, O.V.; Brindley, D.N.; Garcia, J.G. Sphingosine 1-phosphate released from platelets during clotting accounts for the potent endothelial cell chemotactic activity of blood serum and provides a novel link between hemostasis and angiogenesis. FASEB J. 2000, 14, 2255–2265.
- Limaye, V. The role of sphingosine kinase and sphingosine-1-phosphate in the regulation of endothelial cell biology. Endothelium 2008, 15, 101–112.
- Berezin, A.E. Impaired Immune Phenotype of Endothelial Cell-derived Micro Particles: The Missing Link between Diabetes-related States and Risk of Cardiovascular Complications? J. Data Min. Genom. Proteom. 2016, 7, 195–197.
- Li, J.; Salvador, A.M.; Li, G.; Valkov, N.; Ziegler, O.; Yeri, A.; Yang Xiao, C.; Meechoovet, B.; Alsop, E.; Rodosthenous, R.S.; et al. Mir-30d Regulates Cardiac Remodeling by Intracellular and Paracrine Signaling. Circ. Res. 2021, 128, e1–e23.
- He, W.; Huang, H.; Xie, Q.; Wang, Z.; Fan, Y.; Kong, B.; Huang, D.; Xiao, Y. MiR-155 Knockout in Fibroblasts Improves Cardiac Remodeling by Targeting Tumor Protein p53-Inducible Nuclear Protein 1. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 423–435.
- Yuan, J.; Liu, H.; Gao, W.; Zhang, L.; Ye, Y.; Yuan, L.; Ding, Z.; Wu, J.; Kang, L.; Zhang, X.; et al. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress. Theranostics 2018, 8, 2565–2582.
- Schüttler, D.; Clauss, S.; Weckbach, L.T.; Brunner, S. Molecular Mechanisms of Cardiac Remodeling and Regeneration in Physical Exercise. Cells 2019, 8, 1128.
- Maries, L.; Marian, C.; Sosdean, R.; Goanta, F.; Sirbu, I.O.; Anghel, A. MicroRNAs—The Heart of Post-Myocardial Infarction Remodeling. Diagnostics 2021, 11, 1675.
- Wu, Q.; Wang, J.; Tan, W.; Jiang, Y.; Wang, S.; Li, Q.; Yu, X.; Tan, J.; Liu, S.; Zhang, P.; et al. Extracellular vesicles from human embryonic stem cell-derived cardiovascular progenitor cells promote cardiac infarct healing through reducing cardiomyocyte death and promoting angiogenesis. Cell Death Dis. 2020, 11, 354.
- Berezin, A.E.; Berezin, A.A. Extracellular Endothelial Cell-Derived Vesicles: Emerging Role in Cardiac and Vascular Remodeling in Heart Failure. Front. Cardiovasc. Med. 2020, 7, 47.
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A.; et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Investig. 2014, 124, 2136–2146.
- Shaihov-Teper, O.; Ram, E.; Ballan, N.; Brzezinski, R.Y.; Naftali-Shani, N.; Masoud, R.; Ziv, T.; Lewis, N.; Schary, Y.; Levin-Kotler, L.P.; et al. Extracellular Vesicles from Epicardial Fat Facilitate Atrial Fibrillation. Circulation 2021, 143, 2475–2493.