Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Health Care Sciences & Services
Pulmonary arterial hypertension (PAH) is a progressive disease characterized by elevated pulmonary arterial pressure due to increased pulmonary vascular resistance, secondary to sustained pulmonary vasoconstriction and excessive obliterative pulmonary vascular remodeling. Work over the last decade has led to the identification of a critical role for metabolic reprogramming in the PAH pathogenesis. It is becoming clear that in addition to its role in ATP generation, the mitochondrion is an important organelle that regulates complex and integrative metabolic- and signal transduction pathways.
- metabolism
- mitochondria
- pulmonary hypertension
1. Mitochondrial Metabolic Pathways in Pulmonary Hypertension
Recent reports have demonstrated significant alterations in mitochondrial metabolic pathways including glycolysis, glucose oxidation, PPP, glutaminolysis, and FAO. The critical role of mitochondrial metabolic homeostasis in the pathogenesis of PAH is described in detail through these metabolic pathways (Figure 1).
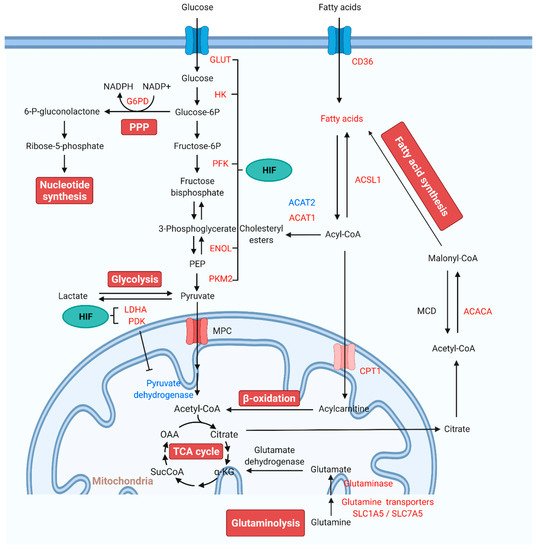
Figure 1. Alterations of mitochondrial metabolic pathways and therapeutic targets for PAH treatment. Under normal conditions, glucose is converted to pyruvate via glycolysis. Pyruvate enters the mitochondria, where it is oxidized in the TCA cycle to generate ATP. In PAH, pyruvate is utilized for lactate production. The production of lactate in the presence of oxygen is known as “aerobic glycolysis” or the Warburg effect. In aerobic glycolysis, excess glucose uptake is diverted through PPP. Glutamine is another fuel source, which enters the mitochondria to replenish TCA intermediates and mobilize cellular energy, carbon, and nitrogen. FAs are the main energy source in the healthy adult heart. FA synthesis is started with the formation of malonyl-CoA by carboxylation of acetyl-CoA. Increased FAO inhibits glucose oxidation. HIF activates the transcription of genes encoding metabolic enzymes that mediate the glycolytic pathway. Red indicates increase, and blue indicates reduction. Abbreviations: ACACA, acetyl-CoA carboxylase; ACAT, acetyl-CoA acetyltransferase; Acetyl-CoA, acetyl coenzyme A; ACSL1, fatty acetyl-CoA L1; Acyl-CoA, acyl-coenzyme A; α-KG, α-ketoglutarate; CPT1, carnitine palmitoyltransferase 1; ENOL, enolase; Fructose-6P, fructose 6-phosphate; Glucose-6P, glucose-6-phosphate; G6PD, glucose-6-phosphate dehydrogenase; GLUT, glucose transporter; HIF, hypoxia-inducible factor; HK, hexokinase; LDHA, lactate dehydrogenase A; MCD, malonyl-CoA decarboxylase; MPC, mitochondrial pyruvate carrier; NADPH, reduced nicotinamide adenine dinucleotide phosphate; OAA, oxaloacetate; 6-P-gluconolactone, 6-phosphate-gluconolactone; PAH, pulmonary arterial hypertension; PDK, pyruvate dehydrogenase kinase; PEP, phosphoenolpyruvate; PFKFB, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase; PKM, pyruvate kinase M; PPP: pentose phosphate pathway; SLC1A5, solute carrier family 1 member 5; SLC7A5, solute carrier family 7 member 5; SucCoA, succinyl-coenzyme A; TCA, tricarboxylic acid.
1.1. Glycolysis and Glucose Oxidation in Pulmonary Arterial Hypertension
Under normal conditions, glycolysis can convert glucose into pyruvate. Pyruvate is transported to mitochondria via mitochondrial pyruvate carrier (MPC) and then oxidized to acetyl coenzyme A (acetyl-CoA) by the pyruvate dehydrogenase (PDH) complex and finally broken down by the TCA cycle. However, in pathological conditions, such as cancer or PAH, pyruvate is converted to lactate by lactate dehydrogenase (LDH) due to PDH inhibition [1]. Positron emission tomography (PET) imaging using fluorine-18-labeled 2-fluoro-2-deoxyglucose (18FDG) shows increased glucose uptake and metabolism in lung tissues and RV of both PAH patients and monocrotaline (MCT)-induced PAH rats, confirming the high glycolytic rate in vivo [2][3][4].
Aerobic glycolysis, a characteristic of non-malignant proliferating cells, has been observed in pulmonary arterial endothelial cells (PAECs) and smooth muscle cells (PASMCs) from idiopathic PAH (IPAH) patients and in rodent PH models [2][5][6]. In IPAH-PAECs, decreased oxygen consumption and increased glycolytic rate underlie the metabolic change from oxidative metabolism to glycolysis in PAH [2]. Less mitochondrial respiration and higher aerobic glycolysis also exists in PASMCs isolated from rats with PH [6]. A similar glycolytic shift has also been observed in RVH [7]. Notably, increased RV 18FDG uptake using PET imaging is associated with the severity of disease as shown by higher RV pressure overload and pulmonary artery (PA) pressures, lower cardiac index, and reduced RV systolic function [8][9]. This relationship between 18FDG uptake and clinical manifestations suggests that metabolic imaging may play a specific role in PAH clinical diagnosis, either as a prognostic marker or to monitor disease progression and treatment effects [10].
PH has striking similarities with cancer. In the endothelial cells (ECs) and SMCs of the pulmonary vascular wall, PH also elicits cellular glycolytic reprogramming, hyperproliferation, and anti-apoptotic phenotypes [2][11][12]. All the above characteristics are similar to those observed in cancer and precisely highlight the Warburg effect, where glycolytic reprogramming provides the necessary energy for the growth and production of new cells [13][14]. Furthermore, the glycolytic intermediates contribute to the production of phospholipids, nucleotides, and amino acids required to sustain cellular replication. This glycolytic reprogramming modifies the vascular cells in the PAs to proliferate excessively, occluding the arteries and restricting blood flow [15][16]. Establishing a link between cancer and PAH has revealed a common reliance upon mitochondrial dysregulation [17][18][19]. Metabolic and mitochondrial dysregulation are key contributors to the development of PAH as mitochondrial hyperpolarization and the glycolytic shift lead to the upregulation of proteins involved in cellular proliferation and downregulation of apoptotic factors. Similar to cancer, the crucial role of mitochondrial function in the cells of the pulmonary vasculature is the metabolic shift to favor glycolysis to produce ATP and lactic acid fermentation.
A global cellular metabolomics overview suggests that a set of altered mitochondrial metabolites contributes to the pathogenesis of PH [20][21]. Studies have revealed that cells of the pulmonary vasculature have a strong dependence on glutamine, which leads to excessive proliferation [22]. In cells, glutamine is the critical amino acid in several biochemical reactions, and is required for glutamate synthesis by glutaminolysis. Further, glutamate is a substrate for glutathione synthesis. Moreover, it is also converted to α-ketoglutarate, which enters the TCA cycle. Overall, Warburg metabolism modifies the cells to depend on glycolysis to produce ATP and cellular building blocks and further inhibits mitochondria-mediated apoptosis, allowing vascular cells to proliferate.
The pathologic accumulation of hypoxia-inducible factor 1α (HIF-1α), an essential transcriptional regulator of the hypoxic response, plays a critical role in regulating the glycolytic shift from mitochondrial oxidation toward aerobic glycolysis in PAH. Researchers have shown that cultured ECs obtained from patients with idiopathic pulmonary arterial hypertension (IPAH-ECs) have a greater HIF-1α expression [23]. Activation of HIF-1α upregulates the transcription of pyruvate dehydrogenase kinase (PDK, a key enzyme of mitochondrial glucose oxidation), which can inhibit the pyruvate dehydrogenase complex (PDH, a regulator of pyruvate uptake into the TCA cycle), suppressing mitochondrial oxidative phosphorylation and increasing aerobic glycolysis [24]. In PASMCs, RV fibroblasts, and RV cardiomyocytes, activation of PDK promotes the metabolic shift, which contributes to disease pathology [25][26][27][28]. Therefore, PDK has been regarded as a promising target for PAH treatment. Administration of a PDK inhibitor dichloroacetate (DCA) in IPAH patients leads to a reduction in mPAP, PVR, and improvement in RV function [29]. Several studies in PAH animal models have also demonstrated that administration of DCA blocks HIF-1α activation and promotes mitochondrial oxidative phosphorylation, thereby preventing and reversing PAH [28][30][31][32]. In addition to the regulation of glycolytic enzymes in response to hypoxia, HIF-1α can upregulate glucose transporter 1 (GLUT1) and hexokinase (HK) to both increase glucose uptake and retain it within the cell, further supporting a glycolytic shift [10][26][33][34]. HIF-1α also activates the transcription of genes encoding pyruvate kinase M 2 (PKM2), lactate dehydrogenase A (LDHA), enolase (ENOL), and other metabolic enzymes that mediate the glycolytic pathway [35][36]. Another central regulator of glycolysis, the mammalian target of rapamycin (mTOR), is also activated, further shifting cellular metabolism to glycolysis [37]. A differential role of mTOR complex 1 (mTORC1) and complex 2 (mTORC2), two functionally distinct mTOR complexes in PAH, has been identified. The disruption of mTORC1 in SMCs ameliorates the development of experimental PAH; however, disruption of mTORC2 leads to spontaneous PAH [38], which indicates that the mTOR signaling pathway is a critical modulator in PAH development. It should be noted that HIF1 and HIF2 can activate overlapping and different genes in different cell types [39], and HIF-2α mediated gene sets differentiate PAH [40]. Importantly, HIF-induced altered glucose metabolism is essential for the production of ATP and limiting mitochondrial ROS (mt-ROS) production. The Warburg effect has been reported in many PAH and RV failure cases and hypoxia-induced PH, leading to the conclusion that pharmacological modification of Warburg effect-mediated pathological phenotype may be a possible strategy to inhibit PAH.
The rate-limiting enzymes in the glycolytic pathway also participate in the glucose metabolic shift in PAH. The 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) catalyzes the conversion of fructose-6-phosphate (F-6-P) to fructose-2,6-bisphosphate (F-2,6-P2), one of three rate-limiting enzymes in glycolysis. PFKFB3 expression is increased in PASMCs and PAECs in both PAH rodents and IPAH patients. Knockdown of PFKFB3 decreases glycolysis, glycolytic capacity, and glycolytic reserve in PVCsfrom IPAH patients. Endothelial PFKFB3 or SMC-specific PFKFB3 deficiency attenuates hypoxia-induced PAH and vascular remodeling. Likewise, PFKFB3 inhibitor 3PO ameliorates MCT- or Sugen 5416/hypoxia (SuHx)-induced pathological changes in hemodynamics, pulmonary vessels, and RV [41][42].
1.2. The Pentose Phosphate Pathway in Pulmonary Arterial Hypertension
Similar to cancer cells, there are ways to increase glucose uptake in PAH through other biosynthetic pathways such as PPP [43]. PPP generates reduced nicotinamide adenine dinucleotide phosphate (NADPH) and ribose-5-phosphate to preserve nucleotide synthesis and redox homeostasis. Increases in PPP flux in PAH patients and multiple animal models have been identified and shown to be associated with the metabolic changes that precede the development of PAH [44][45].
Glucose-6-phosphate dehydrogenase (G6PD) is the first rate-limiting enzyme of the PPP. The increased activity and expression of G6PD have been shown in hypoxic PAs [46], lung tissues of MCT-treated rats [47], endothelin-1 (ET-1)-treated PASMCs [48][49], and hypoxic CD133(+) progenitor cells that maintain cells in high proliferative state [50]. These results indicate that G6PD deficiency may protect against the development of PAH. Indeed, inhibition and knockdown of G6PD in chronic hypoxia-induced PAH and SuHx-PH rat models reverses the metabolic changes, epigenetic modification (DNA methylation), maladaptive expression of genes that contribute to PA remodeling, the formation of occlusive lesions, and RV pressure overload [50][51][52][53]. Protein kinase G1 (PKG1) signaling has a vital role in this process, as G6PD inhibition evokes PKG1α-dependent signaling, thereby mediating expression of contractile proteins, reducing Ang II-induced contraction and proinflammatory factor in PA response to hypoxia [46][54][55]. Inhibition or silencing of G6PD activity induces relaxation of pulmonary and coronary arteries. It attenuates acute HPV via regulating Ca2+ signaling [46] and opening of voltage-gated K+ (Kv) channels [56], indicating a positive linear relationship between G6PD activity and HPV [54]. In addition, a deficiency in G6PD activity in the African, Middle East, and Asian populations has been associated with increased susceptibility to hemolysis [57]. Consistently, PAH patients have significantly decreased G6PD expression and activity, indicating that it may be involved in PAH pathobiology due to increased hemolysis [58].
NADPH is a cofactor for the critical antioxidant enzymes glutathione reductase and thioredoxin reductase, which catalyze the conversion of oxidized glutathione (GSH) and thioredoxins (TRX) to their reduced forms, respectively. NADPH is also a critical cofactor for nitric oxide synthase (NOS), maintaining NO synthesis, and, for NADPH oxidase (NOX) enzymes, generating ROS. The activity of NADPH, NADPH/NADP+ ratio, and NADPH levels are increased in pulmonary microvascular endothelial cells (PMVEC), containing a bone morphogenetic protein receptor type 2 (BMPR2) mutation [45]. A similar phenotype is observed in PASMC [46] and hypoxic PA and lungs [59]. Overproduction of NADPH can cause damaging “reductive stress” in the cardiovascular system [60]. Increases in NADPH levels inhibit redox activation of PKG in the PA in response to hypoxia, which supports phenotypic modulation of PASMCs from a contractile to proliferative phenotype [46]. Meanwhile, generation or maintenance of cytosolic NADPH by G6PD regulates PA vasomotor tone and HPV via inhibition of Kv channels and maintenance of the NO-soluble guanylate cyclase (sGC) pathway [59]. In addition, under pathological conditions, including PAH, the activity of NOXs can be enhanced via utilizing NADPH as a substrate, thereby generating superoxide and hydrogen peroxide (H2O2) [61]. These products participate in chronic obstructive pulmonary disease (COPD)-related and MCT- and hypoxia-induced vascular remodeling [62][63][64] and vasoconstrictor responsiveness [65].
1.3. Glutaminolysis in Pulmonary Arterial Hypertension
Glutaminolysis involves deamination of glutamine to glutamate via glutaminase (GLS) and subsequent conversion of glutamate to α-ketoglutarate (α-KG) by glutamate dehydrogenase. Glutaminolysis contributes to the anaplerosis reaction, whereby the carbon intermediates of the TCA cycle are replenished, and allow cellular energy, and carbon and nitrogen mobilization, especially in the hyperproliferative cells [66].
Increased glutaminolysis in PAH has been found to produce substrate to meet the high energy requirement in hyperproliferative and antiapoptotic PVCs. Specifically, in PVCs and lung tissue of various PAH types, vascular extracellular matrix (ECM) stiffening, an early and potent pathogenic trigger, leads to the yes-associated protein 1 (YAP) and transcriptional coactivator with PDZ binding motif 1 (TAZ) mechanoactivation. Their activation upregulates metabolic enzymes, including GLS1, resulting in glutaminolysis and glycolysis. Anaplerotic production of glutamate and aspartate via GLS1 sustains vascular cell proliferation and migration in a stiff ECM. GLS1-dependent inhibition of glutaminolysis with pharmacologic inhibitors decreased PVC proliferation in vivo and improved manifestations of PAH [67]. Augmented glutaminolysis in PAH promotes lung fibrosis. Glutaminolysis-induced collagen translation and stability by α-KG-mediated mTORC1 activation and collagen proline hydroxylation trigger vascular fibrosis and stiffening, respectively [68]. Pulmonary vascular remodeling is also driven by increased glutaminolysis and glutamate production.
Increased glutaminolysis has been observed in MCT-induced RVH and associated with the maladaptive RVH. The glutaminolysis inhibitor 6-diazo-5-oxo-l-nor-leucine (DON) reduced RVH, and improved cardiac function and treadmill distance in MCT-induced RVH. Moreover, elevated glutamine transporters (SLC1A5 and SLC7A5) suggest an increased glutamine uptake in MCT-induced RVH [69]. Thus, targeting abnormal glutamine metabolism represents a promising therapeutic intervention in treating various forms of PAH.
1.4. Altered Fatty Acid Oxidation in Pulmonary Arterial Hypertension
FAO is a primary cellular energy source in normal adult hearts, supplying approximately 60–90% of ATP for contractile function. The remainder is provided by carbohydrates (glucose and lactate) and ketone bodies oxidation [70]. Various cardiac pathological conditions can disrupt FA metabolism, which contribute to a decrease in cardiac efficiency, contractile dysfunction, and hypertrophy [71][72][73][74]. Increased circulating free FAs and RV lipid deposition in the form of long-chain FAs, triglycerides, diacylglycerols, and ceramides associated with lipotoxic cardiac steatosis as well as increased FAO have been observed in PAH patients [8][71][75][76]. Impaired RVH and triglyceride and ceramide deposition are found in Bmpr2 mutant mice and PA banded rats [71][77]. Increased expression and redistribution of CD36, a FA transporter responsible for FAs uptake into sarcolemma from the intracellular compartment, has been identified in the RVs and cardiomyocytes of the Bmpr2 mutant mouse [78].
Metabolomic results have identified abnormal FA metabolism and accumulation in the pulmonary vasculature in tissues of PAH patients. In particular, dicarboxylic FAs such as tetradecanedioate, hexadecanedioate, and octadecanedioate are found. Moreover, expression of aldehyde dehydrogenase (ALDH), a key enzyme of ω-oxidation, is higher in lung tissues, SMCs, and ECs from patients with PAH, suggesting that ω-oxidation is the main FA oxidation metabolic pathway when β-oxidation is no longer sufficient to supply energy for the pulmonary vascular remodeling in PAH. Several genes encoding enzymes that are involved in FAO, such as fatty acetyl CoA L1 (ACSL1), acyl-CoA dehydrogenase (ACADM), acetyl-CoA acetyltransferase 1 (ACAT1), and acetyl-CoA carboxylase (ACACA), are increased, and ACAT2 expression is decreased in the lungs of PAH patients [79][80]. Importantly, inhibition of FAO may have a beneficial effect in preventing PAH pathogenesis. Lack of malonyl-CoA decarboxylase (MCD) inhibits FAO and converts the metabolic balance to glucose oxidation in vascular media. In animal models, MCD deficiency does not lead to HPV and prevents the development of chronic hypoxia-induced PAH. Trimetazidine and DCA that mimic MCD deletion reduce mPAP, RV hypertrophy, and vascular remodeling, and reverse PAH induced by hypoxia or MCT [81]. Additionally, carnitine palmitoyltransferase 1 (CPT1), a rate-limiting enzyme responsible for transporting acylcarnitine into the mitochondria in the FAO metabolism, is also increased in rat lungs and PAs of the MCT-induced PH model. Enhanced CPT1-driven FAO increased ATP production in lung tissues and promoted PASMC proliferation. At the same time, inhibition of CPT1 activity decreased the level of free FAs entering the mitochondria for FAO, thereby reducing the abnormal accumulation of PASMC [82]. Thus, FA metabolism represents a therapeutic target to treat PAH.
2. Redox Homeostasis in Pulmonary Hypertension
ROS, including hydroxyl radicals, superoxide, and H2O2, exert a crucial role in preserving redox homeostasis [83]. Several enzyme systems regulate ROS formation in the vasculature, including NOXs [84]. Initially, NADPH represents a pivotal reducing equivalent, which is utilized as a substrate to promote NOX activity, thus contributing to pro-oxidant reactions. In addition to the role of the NOX4 subtype in producing H2O2, vascular NOXs release large amounts of superoxide by catalyzing the transfer of single electrons from NADPH to molecular oxygen [85]. NOX homologs are differentially expressed in vascular ECs, SMCs, fibroblasts, or perivascular adipocytes [86]. NOXs-derived ROS has a regulatory role in the pathophysiological function of pulmonary vessels. NOX1 and NOX2 have been reported to contribute to the progression of endothelial dysfunction, inflammation, and hypertension [87][88]. Abnormal expression and activity of NOX4 can cause oxidative stress, senescence, aortic stiffening, and endothelial dysfunction [89][90]. Calcium-dependent NOX5 has been implicated in angiogenesis, vascular remodeling, and calcification [91][92].
As a primary source of ROS, mt-ROS exert an essential role in PAH due to their involvement in apoptosis, inflammation, cell signaling, and mitochondrial DNA (mtDNA) damage. Aberrant antioxidants and mt-ROS production are present in PAH upon the conversion to aerobic glycolysis [93]. In PAs and PASMCs of fawn hooded rat (FHR)-PH, depressed mt-ROS and decreased superoxide dismutase 2 (SOD2, an intramitochondrial antioxidant enzyme) have been correlated with normoxic HIF-1α activation and inhibition of oxygen-sensitive voltage-gated K+ channel expression [30]. Conversely, in PAECs from persistent PAH of the newborn (PPHN), mitochondrially localized manganese superoxide dismutase (MnSOD) expression and activity are inhibited, contributing to oxidative stress and endothelial NO synthase (eNOS) dysfunction. MnSOD transduction of PPHN-PAECs decreases mitochondrial superoxide generation and improves eNOS function and the PA relaxation response [94]. Increased mitochondrial superoxide production is observed in the chronic hypoxia-induced PAH model, triggering HIF-1α stabilization, metabolic reprogramming, and increased intracellular calcium concentration, thereby triggering HPV [93][95][96]. Pharmacological inhibition of MAO-A improves RV afterload and pulmonary vascular remodeling in SuHx rats by reducing pulmonary vascular proliferation and oxidative stress [97]. In addition, accumulating evidence indicates that mt-ROS, NOXs, and other sources of ROS act in concert to stimulate oxidation, which is further facilitated by reduced antioxidant ability (SOD, catalase, glutathione S-transferase, glutathione peroxidase, and thioredoxin) in PVCs in PH (Figure 2) [98][99].
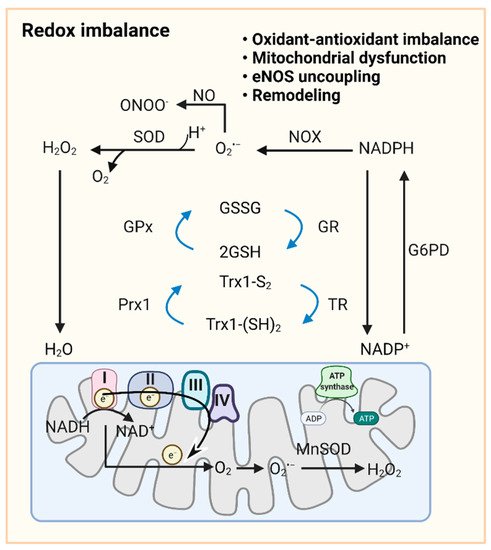
Figure 2. Redox signaling in pulmonary vasculature. GSH and Trx1-(SH)2 are oxidized to GSSG and Trx-S2, recycled in the redox cycle via the NADPH-dependent enzyme GR or TR, while NADP+ can be reduced to NADPH by G6PD in the cytoplasm. Once O2•− is formed by cytoplasmic NOXs and mitochondrial ETC, cytoplasmic SOD and mitochondrial MnSOD catalyze its dismutation into H2O2. GPx uses GSH to further reduce H2O2 to H2O. NO reacts with O2•− to form ONOO−. In mitochondria, ~0.1~0.2% of the total oxygen accepts electrons from ETC to form O2•−. H2O2 is formed by the conversion of O2•− catalyzed by MnSOD or spontaneous dismutation. The production of excessive superoxide free radicals can lead to redox imbalance, mitochondrial damage, uncoupling of eNOS, and ultimately to impaired PAdiastolic function and remodeling. Abbreviations: ADP, adenosine diphosphate; ATP, adenosine triphosphate; eNOS, endothelial NO synthase; ETC, electron transport chain; G6PD, glucose-6-phosphate dehydrogenase; GPx, glutathione peroxidase; GR, glutathione reductase; GSH, reduced glutathione; GSSG, oxidized glutathione; H2O2, hydrogen peroxide; MnSOD, manganese superoxide dismutase; NAD(P)+/NAD(P)H, nicotinamide adenine dinucleotide (phosphate); NO, nitric oxide; NOX, NADPH oxidase; O2•−, superoxide anion; Prx1, peroxiredoxin 1; ONOO−, peroxynitrite; ROS, reactive oxygen species; SOD, superoxide dismutase; TR, thioredoxin reductase; Trx-S2, oxidized thioredoxin; Trx-(SH)2, reduced thioredoxin.
3. Ferroptosis and Lipid Peroxidation in Pulmonary Hypertension
Ferroptosis is a form of regulated cell death induced by the oxidative disturbance of the microenvironment within the cell. This microenvironment is controlled by GPX4 and can be restrained by lipophilic antioxidants and iron chelators. Bioinformatic analysis in lung samples from IPAH patients found the activation of the ferroptosis pathway [100][101]. Administration of deferoxamine, an iron chelator, prevents the development of PH and pulmonary vascular remodeling in rats under hypoxic exposure. In vitro experiments demonstrate that various iron chelators suppress ET-1, platelet-derived growth factor (PDGF), and FBS-induced PASMC proliferation [102]. However, the precise pathological role of ferroptosis in PH development remains unclear.
Lipid peroxidation is evidence for oxidative stress in PH. Samples from PAH patients display increased lipid peroxidation. Specifically, levels of F2-isoprostane, a specific lipid peroxidation product found in the urine of patients with PH, are 2.3 times that of healthy control groups. Changes in mPAP and PVR after NO inhalation are correlated with basal F2-isoprostane levels [103]. There are also research reports demonstrating that urinary F2-isoprostane levels are independently related to the mortality of PAH patients [104]. BMPR2 mutated transgenic mice show a pronounced increase in isoprostanes [105]. Isoprostanes act on the pulmonary vasculature in a variety of ways, including pulmonary vasoconstriction [106], the release of ET-1 [107], and hypertrophy in smooth muscle [108]. Thus, they play an essential mediator role in PH pathologies. Likewise, the level of malonic dialdehyde, an end product of lipid peroxidation in the plasma from IPAH patients, is higher than in healthy volunteers [109]. Together, these studies indicate that PH is causally linked to enhanced lipid peroxidation.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10020341
References
- Chen, Z.; Liu, M.; Li, L.; Chen, L. Involvement of the Warburg effect in non-tumor diseases processes. J. Cell. Physiol. 2018, 233, 2839–2849.
- Xu, W.; Koeck, T.; Lara, A.R.; Neumann, D.; Difilippo, F.P.; Koo, M.; Janocha, A.J.; Masri, F.A.; Arroliga, A.C.; Jennings, C.; et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl. Acad. Sci. USA 2007, 104, 1342–1347.
- Hagan, G.; Southwood, M.; Treacy, C.; Ross, R.M.; Soon, E.; Coulson, J.; Sheares, K.; Screaton, N.; Pepke-Zaba, J.; Morrell, N.W.; et al. 18FDG PET imaging can quantify increased cellular metabolism in pulmonary arterial hypertension: A proof-of-principle study. Pulm. Circ. 2011, 1, 448–455.
- Zhao, L.; Ashek, A.; Wang, L.; Fang, W.; Dabral, S.; Dubois, O.; Cupitt, J.; Pullamsetti, S.S.; Cotroneo, E.; Jones, H.; et al. Heterogeneity in lung 18FDG uptake in pulmonary arterial hypertension: Potential of dynamic 18FDG positron emission tomography with kinetic analysis as a bridging biomarker for pulmonary vascular remodeling targeted treatments. Circulation 2013, 128, 1214–1224.
- Hernandez-Saavedra, D.; Sanders, L.; Freeman, S.; Reisz, J.A.; Lee, M.H.; Mickael, C.; Kumar, R.; Kassa, B.; Gu, S.; D’Alessandro, A.; et al. Stable isotope metabolomics of pulmonary artery smooth muscle and endothelial cells in pulmonary hypertension and with TGF-beta treatment. Sci. Rep. 2020, 10, 413.
- Rafikov, R.; Sun, X.; Rafikova, O.; Louise Meadows, M.; Desai, A.A.; Khalpey, Z.; Yuan, J.X.; Fineman, J.R.; Black, S.M. Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox Biol. 2015, 6, 278–286.
- Piao, L.; Marsboom, G.; Archer, S.L. Mitochondrial metabolic adaptation in right ventricular hypertrophy and failure. J. Mol. Med. 2010, 88, 1011–1020.
- Ohira, H.; Dekemp, R.; Pena, E.; Davies, R.A.; Stewart, D.J.; Chandy, G.; Contreras-Dominguez, V.; Dennie, C.; Mc Ardle, B.; Mc Klein, R.; et al. Shifts in myocardial fatty acid and glucose metabolism in pulmonary arterial hypertension: A potential mechanism for a maladaptive right ventricular response. Eur. Heart J. Cardiovasc. Imaging 2016, 17, 1424–1431.
- Bokhari, S.; Raina, A.; Rosenweig, E.B.; Schulze, P.C.; Bokhari, J.; Einstein, A.J.; Barst, R.J.; Johnson, L.L. PET imaging may provide a novel biomarker and understanding of right ventricular dysfunction in patients with idiopathic pulmonary arterial hypertension. Circ. Cardiovasc. Imaging 2011, 4, 641–647.
- Marsboom, G.; Wietholt, C.; Haney, C.R.; Toth, P.T.; Ryan, J.J.; Morrow, E.; Thenappan, T.; Bache-Wiig, P.; Piao, L.; Paul, J.; et al. Lung ¹⁸F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 670–679.
- Rehman, J.; Archer, S.L. A proposed mitochondrial-metabolic mechanism for initiation and maintenance of pulmonary arterial hypertension in fawn-hooded rats: The Warburg model of pulmonary arterial hypertension. Adv. Exp. Med. Biol. 2010, 661, 171–185.
- Xu, W.; Erzurum, S.C. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Compr. Physiol. 2011, 1, 357–372.
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Shi, J.; Yang, Y.; Cheng, A.; Xu, G.; He, F. Metabolism of vascular smooth muscle cells in vascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H613–H631.
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887.
- Lahera, V.; De Las Heras, N.; López-Farré, A.; Manucha, W.; Ferder, L. Role of Mitochondrial Dysfunction in Hypertension and Obesity. Curr. Hypertens. Rep. 2017, 19, 11.
- Eirin, A.; Lerman, A.; Lerman, L.O. Mitochondrial injury and dysfunction in hypertension-induced cardiac damage. Eur. Heart J. 2014, 35, 3258–3266.
- Archer, S.L.; Gomberg-Maitland, M.; Maitland, M.L.; Rich, S.; Garcia, J.G.N.; Weir, E.K. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ROS-HIF-1α-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am. J. Physiol.-Heart Circ. Physiol. 2008, 294, H570–H578.
- Rhodes, C.J.; Ghataorhe, P.; Wharton, J.; Rue-Albrecht, K.C.; Hadinnapola, C.; Watson, G.; Bleda, M.; Haimel, M.; Coghlan, G.; Corris, P.A.; et al. Plasma Metabolomics Implicates Modified Transfer RNAs and Altered Bioenergetics in the Outcomes of Pulmonary Arterial Hypertension. Circulation 2017, 135, 460–475.
- Bujak, R.; Mateo, J.; Blanco, I.; Izquierdo-Garcia, J.L.; Dudzik, D.; Markuszewski, M.J.; Peinado, V.I.; Laclaustra, M.; Barbera, J.A.; Barbas, C.; et al. New Biochemical Insights into the Mechanisms of Pulmonary Arterial Hypertension in Humans. PLoS ONE 2016, 11, e0160505.
- Mprah, R.; Adzika, G.K.; Gyasi, Y.I.; Ndzie Noah, M.L.; Adu-Amankwaah, J.; Adekunle, A.O.; Duah, M.; Wowui, P.I.; Weili, Q. Glutaminolysis: A Driver of Vascular and Cardiac Remodeling in Pulmonary Arterial Hypertension. Front. Cardiovasc. Med. 2021, 8, 667446.
- Fijalkowska, I.; Xu, W.; Comhair, S.a.A.; Janocha, A.J.; Mavrakis, L.A.; Krishnamachary, B.; Zhen, L.; Mao, T.; Richter, A.; Erzurum, S.C.; et al. Hypoxia Inducible-Factor1α Regulates the Metabolic Shift of Pulmonary Hypertensive Endothelial Cells. Am. J. Pathol. 2010, 176, 1130–1138.
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197.
- Plecita-Hlavata, L.; Tauber, J.; Li, M.; Zhang, H.; Flockton, A.R.; Pullamsetti, S.S.; Chelladurai, P.; D’alessandro, A.; El Kasmi, K.C.; Jezek, P.; et al. Constitutive Reprogramming of Fibroblast Mitochondrial Metabolism in Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2016, 55, 47–57.
- Piao, L.; Fang, Y.H.; Cadete, V.J.; Wietholt, C.; Urboniene, D.; Toth, P.T.; Marsboom, G.; Zhang, H.J.; Haber, I.; Rehman, J.; et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: Resuscitating the hibernating right ventricle. J. Mol. Med. 2010, 88, 47–60.
- Li, B.; Zhu, Y.; Sun, Q.; Yu, C.; Chen, L.; Tian, Y.; Yan, J. Reversal of the Warburg effect with DCA in PDGF-treated human PASMC is potentiated by pyruvate dehydrogenase kinase-1 inhibition mediated through blocking Akt/GSK-3β signalling. Int. J. Mol. Med. 2018, 42, 1391–1400.
- Tian, L.; Wu, D.; Dasgupta, A.; Chen, K.H.; Mewburn, J.; Potus, F.; Lima, P.D.A.; Hong, Z.; Zhao, Y.Y.; Hindmarch, C.C.T.; et al. Epigenetic Metabolic Reprogramming of Right Ventricular Fibroblasts in Pulmonary Arterial Hypertension: A Pyruvate Dehydrogenase Kinase-Dependent Shift in Mitochondrial Metabolism Promotes Right Ventricular Fibrosis. Circ. Res. 2020, 126, 1723–1745.
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9, eaao4583.
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thébaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; Mcmurtry, M.S.; et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641.
- Liu, P.; Huang, W.; Ding, Y. Fasudil Dichloroacetate Alleviates SU5416/Hypoxia-Induced Pulmonary Arterial Hypertension by Ameliorating Dysfunction of Pulmonary Arterial Smooth Muscle Cells. Drug Des. Dev. Ther. 2021, 15, 1653–1666.
- Mcmurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 2004, 95, 830–840.
- Chen, C.; Luo, F.; Wu, P.; Huang, Y.; Das, A.; Chen, S.; Chen, J.; Hu, X.; Li, F.; Fang, Z. Metabolomics reveals metabolite changes of patients with pulmonary arterial hypertension in China. J. Cell. Mol. Med. 2020, 24, 2484–2496.
- Fulda, S.; Debatin, K.M. HIF-1-regulated glucose metabolism: A key to apoptosis resistance? Cell Cycle 2007, 6, 790–792.
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744.
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol Chem. 1994, 269, 23757–23763.
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684.
- Tang, H.; Wu, K.; Wang, J.; Vinjamuri, S.; Gu, Y.; Song, S.; Wang, Z.; Zhang, Q.; Balistrieri, A.; Ayon, R.J.; et al. Pathogenic Role of mTORC1 and mTORC2 in Pulmonary Hypertension. JACC Basic Transl. Sci. 2018, 3, 744–762.
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 versus HIF-2--is one more important than the other? Vascul. Pharmacol. 2012, 56, 245–251.
- Zhu, J.; Zhao, L.; Hu, Y.; Cui, G.; Luo, A.; Bao, C.; Han, Y.; Zhou, T.; Lu, W.; Wang, J.; et al. Hypoxia-Inducible Factor 2-Alpha Mediated Gene Sets Differentiate Pulmonary Arterial Hypertension. Front. Cell Dev. Biol. 2021, 9, 701247.
- Kovacs, L.; Cao, Y.; Han, W.; Meadows, L.; Kovacs-Kasa, A.; Kondrikov, D.; Verin, A.D.; Barman, S.A.; Dong, Z.; Huo, Y.; et al. PFKFB3 in Smooth Muscle Promotes Vascular Remodeling in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 617–627.
- Cao, Y.; Zhang, X.; Wang, L.; Yang, Q.; Ma, Q.; Xu, J.; Wang, J.; Kovacs, L.; Ayon, R.J.; Liu, Z.; et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2019, 116, 13394–13403.
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354.
- Rafikova, O.; Meadows, M.L.; Kinchen, J.M.; Mohney, R.P.; Maltepe, E.; Desai, A.A.; Yuan, J.X.; Garcia, J.G.; Fineman, J.R.; Rafikov, R.; et al. Metabolic Changes Precede the Development of Pulmonary Hypertension in the Monocrotaline Exposed Rat Lung. PLoS ONE 2016, 11, e0150480.
- Fessel, J.P.; Hamid, R.; Wittmann, B.M.; Robinson, L.J.; Blackwell, T.; Tada, Y.; Tanabe, N.; Tatsumi, K.; Hemnes, A.R.; West, J.D. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm. Circ. 2012, 2, 201–213.
- Chettimada, S.; Rawat, D.K.; Dey, N.; Kobelja, R.; Simms, Z.; Wolin, M.S.; Lincoln, T.M.; Gupte, S.A. Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L64–L74.
- Sun, X.; Kumar, S.; Sharma, S.; Aggarwal, S.; Lu, Q.; Gross, C.; Rafikova, O.; Lee, S.G.; Dasarathy, S.; Hou, Y.; et al. Endothelin-1 induces a glycolytic switch in pulmonary arterial endothelial cells via the mitochondrial translocation of endothelial nitric oxide synthase. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1084–1095.
- Chettimada, S.; Gupte, R.; Rawat, D.; Gebb, S.A.; Mcmurtry, I.F.; Gupte, S.A. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and -activation in pulmonary artery smooth muscle cells: Implication in pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L287–L300.
- Yao, C.; Yu, J.; Taylor, L.; Polgar, P.; Mccomb, M.E.; Costello, C.E. Protein Expression by Human Pulmonary Artery Smooth Muscle Cells Containing a BMPR2 Mutation and the Action of ET-1 as Determined by Proteomic Mass Spectrometry. Int. J. Mass Spectrom. 2015, 378, 347–359.
- Chettimada, S.; Joshi, S.R.; Alzoubi, A.; Gebb, S.A.; Mcmurtry, I.F.; Gupte, R.; Gupte, S.A. Glucose-6-phosphate dehydrogenase plays a critical role in hypoxia-induced CD133+ progenitor cells self-renewal and stimulates their accumulation in the lungs of pulmonary hypertensive rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L545–L556.
- Kitagawa, A.; Jacob, C.; Jordan, A.; Waddell, I.; McMurtry, I.F.; Gupte, S.A. Inhibition of Glucose-6-Phosphate Dehydrogenase Activity Attenuates Right Ventricle Pressure and Hypertrophy Elicited by VEGFR Inhibitor + Hypoxia. J. Pharmacol. Exp. Ther. 2021, 377, 284–292.
- Joshi, S.R.; Kitagawa, A.; Jacob, C.; Hashimoto, R.; Dhagia, V.; Ramesh, A.; Zheng, C.; Zhang, H.; Jordan, A.; Waddell, I.; et al. Hypoxic activation of glucose-6-phosphate dehydrogenase controls the expression of genes involved in the pathogenesis of pulmonary hypertension through the regulation of DNA methylation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L773–L786.
- Varghese, M.V.; James, J.; Rafikova, O.; Rafikov, R. Glucose-6-phosphate dehydrogenase deficiency contributes to metabolic abnormality and pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L508–L521.
- Gupte, R.S.; Rawat, D.K.; Chettimada, S.; Cioffi, D.L.; Wolin, M.S.; Gerthoffer, W.T.; Mcmurtry, I.F.; Gupte, S.A. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J. Biol. Chem. 2010, 285, 19561–19571.
- Lakhkar, A.; Dhagia, V.; Joshi, S.R.; Gotlinger, K.; Patel, D.; Sun, D.; Wolin, M.S.; Schwartzman, M.L.; Gupte, S.A. 20-HETE-induced mitochondrial superoxide production and inflammatory phenotype in vascular smooth muscle is prevented by glucose-6-phosphate dehydrogenase inhibition. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1107–H1117.
- Gupte, S.A.; Li, K.X.; Okada, T.; Sato, K.; Oka, M. Inhibitors of pentose phosphate pathway cause vasodilation: Involvement of voltage-gated potassium channels. J. Pharmacol. Exp. Ther. 2002, 301, 299–305.
- Tzounakas, V.L.; Kriebardis, A.G.; Georgatzakou, H.T.; Foudoulaki-Paparizos, L.E.; Dzieciatkowska, M.; Wither, M.J.; Nemkov, T.; Hansen, K.C.; Papassideri, I.S.; D’alessandro, A.; et al. Glucose 6-phosphate dehydrogenase deficient subjects may be better “storers” than donors of red blood cells. Free Radic. Biol. Med. 2016, 96, 152–165.
- Kurdyukov, S.; Eccles, C.A.; Desai, A.; Gonzalez-Garay, M.; Yuan, J.X.-J.; Garcia, J.G.N.; Rafikova, O.; Rafikov, R. New cases of Glucose-6-Phosphate Dehydrogenase deficiency in Pulmonary Arterial Hypertension. PLoS ONE 2018, 13, e0203493.
- Gupte, S.A.; Okada, T.; Mcmurtry, I.F.; Oka, M. Role of pentose phosphate pathway-derived NADPH in hypoxic pulmonary vasoconstriction. Pulm. Pharmacol. Ther. 2006, 19, 303–309.
- Karimi Galougahi, K.; Ashley, E.A.; Ali, Z.A. Redox regulation of vascular remodeling. Cell Mol. Life Sci. 2016, 73, 349–363.
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890.
- Barman, S.A.; Chen, F.; Su, Y.; Dimitropoulou, C.; Wang, Y.; Catravas, J.D.; Han, W.; Orfi, L.; Szantai-Kis, C.; Keri, G.; et al. NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arter. Thromb. Vasc. Biol. 2014, 34, 1704–1715.
- Li, S.; Tabar, S.S.; Malec, V.; Eul, B.G.; Klepetko, W.; Weissmann, N.; Grimminger, F.; Seeger, W.; Rose, F.; Hänze, J. NOX4 regulates ROS levels under normoxic and hypoxic conditions, triggers proliferation, and inhibits apoptosis in pulmonary artery adventitial fibroblasts. Antioxid. Redox Signal. 2008, 10, 1687–1698.
- Guo, X.; Fan, Y.; Cui, J.; Hao, B.; Zhu, L.; Sun, X.; He, J.; Yang, J.; Dong, J.; Wang, Y.; et al. NOX4 expression and distal arteriolar remodeling correlate with pulmonary hypertension in COPD. BMC Pulm. Med. 2018, 18, 111.
- Liu, J.Q.; Zelko, I.N.; Erbynn, E.M.; Sham, J.S.; Folz, R.J. Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox). Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L2–L10.
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464.
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335.
- Ge, J.; Cui, H.; Xie, N.; Banerjee, S.; Guo, S.; Dubey, S.; Barnes, S.; Liu, G. Glutaminolysis Promotes Collagen Translation and Stability via α-Ketoglutarate-mediated mTOR Activation and Proline Hydroxylation. Am. J. Respir. Cell Mol. Biol. 2018, 58, 378–390.
- Piao, L.; Fang, Y.H.; Parikh, K.; Ryan, J.J.; Toth, P.T.; Archer, S.L. Cardiac glutaminolysis: A maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J. Mol. Med. 2013, 91, 1185–1197.
- Van Der Vusse, G.J.; Van Bilsen, M.; Glatz, J.F. Cardiac fatty acid uptake and transport in health and disease. Cardiovasc. Res. 2000, 45, 279–293.
- Hemnes, A.R.; Brittain, E.L.; Trammell, A.W.; Fessel, J.P.; Austin, E.D.; Penner, N.; Maynard, K.B.; Gleaves, L.; Talati, M.; Absi, T.; et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 325–334.
- Graham, B.B.; Kumar, R.; Mickael, C.; Sanders, L.; Gebreab, L.; Huber, K.M.; Perez, M.; Smith-Jones, P.; Serkova, N.J.; Tuder, R.M. Severe pulmonary hypertension is associated with altered right ventricle metabolic substrate uptake. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L435–L440.
- Tuunanen, H.; Engblom, E.; Naum, A.; Någren, K.; Hesse, B.; Airaksinen, K.E.; Nuutila, P.; Iozzo, P.; Ukkonen, H.; Opie, L.H.; et al. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation 2006, 114, 2130–2137.
- Lopaschuk, G.D. Targets for modulation of fatty acid oxidation in the heart. Curr. Opin. Investig. Drugs. 2004, 5, 290–294.
- Brittain, E.L.; Talati, M.; Fessel, J.P.; Zhu, H.; Penner, N.; Calcutt, M.W.; West, J.D.; Funke, M.; Lewis, G.D.; Gerszten, R.E.; et al. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 2016, 133, 1936–1944.
- Sakao, S.; Miyauchi, H.; Voelkel, N.F.; Sugiura, T.; Tanabe, N.; Kobayashi, Y.; Tatsumi, K. Increased Right Ventricular Fatty Acid Accumulation in Chronic Thromboembolic Pulmonary Hypertension. Ann. Am. Thorac. Soc. 2015, 12, 1465–1472.
- Fang, Y.H.; Piao, L.; Hong, Z.; Toth, P.T.; Marsboom, G.; Bache-Wiig, P.; Rehman, J.; Archer, S.L. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: Exploiting Randle’s cycle. J. Mol. Med. 2012, 90, 31–43.
- Talati, M.H.; Brittain, E.L.; Fessel, J.P.; Penner, N.; Atkinson, J.; Funke, M.; Grueter, C.; Jerome, W.G.; Freeman, M.; Newman, J.H.; et al. Mechanisms of Lipid Accumulation in the Bone Morphogenetic Protein Receptor Type 2 Mutant Right Ventricle. Am. J. Respir. Crit. Care Med. 2016, 194, 719–728.
- Zhao, Y.; Peng, J.; Lu, C.; Hsin, M.; Mura, M.; Wu, L.; Chu, L.; Zamel, R.; Machuca, T.; Waddell, T.; et al. Metabolomic heterogeneity of pulmonary arterial hypertension. PLoS ONE 2014, 9, e88727.
- Xu, W.; Comhair, S.A.A.; Chen, R.; Hu, B.; Hou, Y.; Zhou, Y.; Mavrakis, L.A.; Janocha, A.J.; Li, L.; Zhang, D.; et al. Integrative proteomics and phosphoproteomics in pulmonary arterial hypertension. Sci. Rep. 2019, 9, 18623.
- Sutendra, G.; Bonnet, S.; Rochefort, G.; Haromy, A.; Folmes, K.D.; Lopaschuk, G.D.; Dyck, J.R.; Michelakis, E.D. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci. Transl. Med. 2010, 2, 44ra58.
- Zhuang, W.; Lian, G.; Huang, B.; Du, A.; Gong, J.; Xiao, G.; Xu, C.; Wang, H.; Xie, L. CPT1 regulates the proliferation of pulmonary artery smooth muscle cells through the AMPK-p53-p21 pathway in pulmonary arterial hypertension. Mol. Cell. Biochem. 2018, 455, 169–183.
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990.
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120.
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814.
- Muñoz, M.; López-Oliva, M.E.; Rodríguez, C.; Martínez, M.P.; Sáenz-Medina, J.; Sánchez, A.; Climent, B.; Benedito, S.; García-Sacristán, A.; Rivera, L.; et al. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol. 2020, 28, 101330.
- García-Redondo, A.B.; Aguado, A.; Briones, A.M.; Salaices, M. NADPH oxidases and vascular remodeling in cardiovascular diseases. Pharmacol. Res. 2016, 114, 110–120.
- Salazar, G. NADPH Oxidases and Mitochondria in Vascular Senescence. Int. J. Mol. Sci. 2018, 19, 1327.
- Canugovi, C.; Stevenson, M.D.; Vendrov, A.E.; Hayami, T.; Robidoux, J.; Xiao, H.; Zhang, Y.Y.; Eitzman, D.T.; Runge, M.S.; Madamanchi, N.R. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. 2019, 26, 101288.
- Furmanik, M.; Chatrou, M.; Van Gorp, R.; Akbulut, A.; Willems, B.; Schmidt, H.; Van Eys, G.; Bochaton-Piallat, M.L.; Proudfoot, D.; Biessen, E.; et al. Reactive Oxygen-Forming Nox5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ. Res. 2020, 127, 911–927.
- Montezano, A.C.; Tsiropoulou, S.; Dulak-Lis, M.; Harvey, A.; Camargo Lde, L.; Touyz, R.M. Redox signaling, Nox5 and vascular remodeling in hypertension. Curr. Opin. Nephrol. Hypertens. 2015, 24, 425–433.
- Archer, S.L. Acquired Mitochondrial Abnormalities, Including Epigenetic Inhibition of Superoxide Dismutase 2, in Pulmonary Hypertension and Cancer: Therapeutic Implications. Adv. Exp. Med. Biol. 2016, 903, 29–53.
- Afolayan, A.J.; Eis, A.; Teng, R.J.; Bakhutashvili, I.; Kaul, S.; Davis, J.M.; Konduri, G.G. Decreases in manganese superoxide dismutase expression and activity contribute to oxidative stress in persistent pulmonary hypertension of the newborn. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L870–L879.
- Waypa, G.B.; Marks, J.D.; Guzy, R.D.; Mungai, P.T.; Schriewer, J.M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary Circulation. Am. J. Respir. Crit. Care Med. 2013, 187, 424–432.
- Chi, A.Y.; Waypa, G.B.; Mungai, P.T.; Schumacker, P.T. Prolonged hypoxia increases ROS signaling and RhoA activation in pulmonary artery smooth muscle and endothelial cells. Antioxid. Redox Signal. 2010, 12, 603–610.
- Sun, X.Q.; Peters, E.L.; Schalij, I.; Axelsen, J.B.; Andersen, S.; Kurakula, K.; Gomez-Puerto, M.C.; Szulcek, R.; Pan, X.; Da Silva Goncalves Bos, D. Increased MAO-A Activity Promotes Progression of Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2021, 64, 331–343.
- Song, T.; Zheng, Y.M.; Wang, Y.X. Cross Talk Between Mitochondrial Reactive Oxygen Species and Sarcoplasmic Reticulum Calcium in Pulmonary Arterial Smooth Muscle Cells. Adv. Exp. Med. Biol. 2017, 967, 289–298.
- Frazziano, G.; Al Ghouleh, I.; Baust, J.; Shiva, S.; Champion, H.C.; Pagano, P.J. Nox-derived ROS are acutely activated in pressure overload pulmonary hypertension: Indications for a seminal role for mitochondrial Nox4. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H197–H205.
- Zou, H.X.; Qiu, B.Q. Iron Metabolism and Idiopathic Pulmonary Arterial Hypertension: New Insights from Bioinformatic Analysis. Biomed. Res. Int. 2021, 2021, 5669412.
- Zhang, F.; Liu, H. Identification of ferroptosis-associated genes exhibiting altered expression in pulmonary arterial hypertension. Math. Biosci Eng. 2021, 18, 7619–7630.
- Wong, C.M.; Preston, I.R.; Hill, N.S.; Suzuki, Y.J. Iron chelation inhibits the development of pulmonary vascular remodeling. Free Radic. Biol. Med. 2012, 53, 1738–1747.
- Cracowski, J.L.; Cracowski, C.; Bessard, G.; Pepin, J.L.; Bessard, J.; Schwebel, C.; Stanke-Labesque, F.; Pison, C. Increased lipid peroxidation in patients with pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2001, 164, 1038–1042.
- Cracowski, J.L.; Degano, B.; Chabot, F.; Labarère, J.; Schwedhelm, E.; Monneret, D.; Iuliano, L.; Schwebel, C.; Chaouat, A.; Reynaud-Gaubert, M.; et al. Independent association of urinary F2-isoprostanes with survival in pulmonary arterial hypertension. Chest 2012, 142, 869–876.
- Lane, K.L.; Talati, M.; Austin, E.; Hemnes, A.R.; Johnson, J.A.; Fessel, J.P.; Blackwell, T.; Mernaugh, R.L.; Robinson, L.; Fike, C.; et al. Oxidative injury is a common consequence of BMPR2 mutations. Pulm. Circ. 2011, 1, 72–83.
- Janssen, L.J.; Premji, M.; Netherton, S.; Coruzzi, J.; Lu-Chao, H.; Cox, P.G. Vasoconstrictor actions of isoprostanes via tyrosine kinase and Rho kinase in human and canine pulmonary vascular smooth muscles. Br. J. Pharmacol. 2001, 132, 127–134.
- Yi, S.L.; Kantores, C.; Belcastro, R.; Cabacungan, J.; Tanswell, A.K.; Jankov, R.P. 8-Isoprostane-induced endothelin-1 production by infant rat pulmonary artery smooth muscle cells is mediated by Rho-kinase. Free Radic. Biol. Med. 2006, 41, 942–949.
- Katsuyama, M.; Fan, C.; Yabe-Nishimura, C. NADPH oxidase is involved in prostaglandin F2alpha-induced hypertrophy of vascular smooth muscle cells: Induction of NOX1 by PGF2alpha. J. Biol. Chem. 2002, 277, 13438–13442.
- Irodova, N.L.; Lankin, V.Z.; Konovalova, G.K.; Kochetov, A.G.; Chazova, I.E. Oxidative stress in patients with primary pulmonary hypertension. Bull. Exp. Biol Med. 2002, 133, 580–582.
This entry is offline, you can click here to edit this entry!