Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Non-alcoholic fatty liver disease (NAFLD), which approximately affects a quarter of the world’s population, has become a major public health concern. Although usually associated with excess body weight, it may also affect normal-weight individuals, a condition termed as lean/non-obese NAFLD. The prevalence of lean/non-obese NAFLD is around 20% within the NAFLD population, and 5% within the general population. Current treatment of lean NAFLD is aimed at improving overall fitness and decreasing visceral adiposity, with weight loss strategies being the cornerstone of treatment.
- lean NAFLD
- visceral adiposity
- insulin resistance
- gut microbiota
- metabolic syndrome
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is a spectrum of liver conditions, ranging from (1) simple steatosis (non-alcoholic fatty liver; NAFL), with a low risk of progression; (2) non-alcoholic steatohepatitis (NASH), associated with inflammation and hepatocellular injury (characterized histologically by ballooning); and (3) advanced liver fibrosis, associated with an increased likelihood of progressing to cirrhosis and a higher risk of liver-related mortality [1][2]. Despite the fact that NAFLD has been increasing in prevalence over the past 2 decades, in parallel with the rising prevalence of obesity, it has also been noted that the prevalence of NAFLD is increasing in individuals with normal weight (defined by a body mass index, BMI <25 kg/m2 in Caucasians and a BMI <23 kg/m2 in Asians), a condition that has been defined as lean NAFLD [1]. Moreover, some studies have also coined the term non-obese NAFLD, which includes individuals with a BMI <30 kg/m2 in the Caucasian population and a BMI <25 kg/m2 in the Asian population [3][4]
According to epidemiological studies, approximately 10–20% of individuals with a diagnosis of NAFLD are lean [5]. Thus, in the United States, lean NAFLD is estimated to affect about 8 million–10 million individuals [6]. Importantly, lean NAFLD is not a benign condition, as it can progress to a more severe liver disease such as NASH and advanced fibrosis, which can further lead to cirrhosis [1]. Moreover, a number of studies indicate that individuals with lean NAFLD have an increased risk of developing type 2 diabetes mellitus (T2DM) and have increased all-cause mortality, as compared with subjects with obesity and NAFLD [6][7].
Taking all these data together, developing strategies to identify high-risk patients of developing lean NAFLD and designing effective therapeutic approaches for this condition should be considered as a health priority, since lean NAFLD, unfortunately, may go unnoticed for years, due to the absence of clinical manifestations, and be undetected until stages in which hepatic damage is advanced and the prognosis can be compromised.
2. Prevalence of Lean/Non-Obese NAFLD
The prevalence rates of lean/non-obese NAFLD vary widely, ranging from 3% to 30% in the world population. This variability may be attributed to several factors such as patient selection, diagnostic modalities, BMI cut-off values, and lifestyle and dietary customs of the evaluated populations [6].
In a study from the United States, in which the prevalence of NAFLD was estimated using data from the National Health and Nutritional Examination Survey III (NHANES III) database (1988–1991), Younossi et al. found that, among 11,613 eligible participants, 18.8% had NAFLD and 3.7% had lean NAFLD [7]. The overall prevalence of NAFLD among lean subjects was 9.7% (431/4457), whereas it was 28.8% (2061/7156) in non-lean subjects [7]. However, this is the oldest study that evaluated the prevalence of NAFLD in lean individuals. In a recent study, also from the United States, Zou et al. found that the overall prevalence of NAFLD was 32.3%; among those with NAFLD, 29.7% were non-obese, of which 13.6% had lean NAFLD [8]. In studies conducted on a Korean population, the overall prevalence of NAFLD was 20.1%, with a NAFLD prevalence ranging from 12.6% to 27.4% in non-obese subjects [9][10]. Kim et al. found that in Korean individuals the prevalence of NAFLD was 37.5%, with a lean NAFLD prevalence of 11% [11]. In China, the prevalence of NAFLD was 7.3% in 6905 non-obese participants [12]. In another study, among 1779 Chinese individuals with a BMI < 24 kg/m2, Feng et al. found that 7.5% of individuals had ultrasound-detected liver steatosis [13]. In Hong Kong, the prevalence rate of NAFLD based on proton-MRI spectroscopy (1H-MRS) was 14.8% in non-obese individuals [14]. In a study from Japan, Nishioji et al. found that the prevalence rate of non-obese NAFLD was 12.6% [15]. Additionally, in a biopsy-based study, among 157 lean liver donors from India, 53 (33.7%) had NAFLD [16]. A large meta-analysis of 84 studies showed that within the NAFLD population, 19.2% of subjects were lean and 40.8% were non-obese [17]. The same meta-analysis reported that, in the general population (comprising individuals with and without NAFLD), 12.1% of people had non-obese NAFLD and 5.1% had lean NAFLD [17]. A meta-analysis of 55,936 lean/non-obese individuals by Shi et al. reported that the pooled prevalence of NAFLD was 10.2% and 15.7% in the lean and non-obese population, respectively [18]. Zou et al. reported in a meta-analysis that included 155,846 non-obese participants an overall prevalence of NAFLD of 14.5% [19]. Finally, a meta-analysis of 205,307 individuals from 14 countries reported 4.1% as the global prevalence of lean NAFLD [20]. Table 1 shows the most relevant epidemiological studies of both lean and non-obese NAFLD, including the method for the NAFL diagnosis.
Table 1. Selected prevalence studies of lean and non-obese NAFLD in adult populations.
| Study, Year | Country | Population | Sample Size | Non-Obese NAFLD * Prevalence (% of Population) |
Lean NAFLD ** Prevalence (% of Population) |
Mode of Diagnosis | Overall NAFLD Prevalence (in Population) |
|---|---|---|---|---|---|---|---|
| Kwon et al., 2012 [10] | Korea | Hospital-based | 29,994 | 12.6% | - | USG | 20.1% |
| Younossi et al., 2012 [7] | USA | NHANES III database (1988–1991) |
11,613 | - | 3.7% | USG | 18.8% |
| Sinn et al., 2012 [9] | Korea | Non-obese population | 5878 | 27.4% | 16% | USG | - |
| Xu et al., 2013 [12] | China | Non-obese population | 6905 | 7.27% | - | USG | - |
| Feng et al., 2014 [13] | China | Annual health check-ups | 1779 | - | 7.5% | USG | 50.5% |
| Nishioji et al., 2015 [15] | Japan | Health check-ups | 3271 | 12.6% | - | USG | 24.6% |
| Wei et al., 2015 [14] | Hong Kong | Urban general population | 911 | 14.8% | - | 1H-MRS | 28.8% |
| Ye et al., 2020 [17] | Global | Global | 10,530,308 | 12.1% | 5.1% | Mainly USG | - |
| Zou et al., 2020 [19] | USA | General population | 14,365 | 9.6% | 1.3% | USG/fatty liver index | 32.3% |
| Lu et al., 2020 [20] | Global | Global | 205,307 | - | 4.1% | Mainly USG | - |
| Kim et al., 2021 [11] | Korea | General, KNHANES (2008–2010) | 4786 | - | 11% | Comprehensive NAFLD score | 37.5% |
| Shi et al., 2020 [18] | China | Lean/non-obese | 55,936 | 15.7% | 10.2% | Mainly USG | - |
* Non-obese is defined as <25 kg/m2 in Asians and <30 kg/m2 in Caucasians. ** Lean is defined as BMI < 23 kg/m2 in Asian populations and <25 kg/m2 in Caucasians. KNHANES: Korean National Health and Nutrition Examination Survey. 1H-MRS: proton magnetic resonance spectroscopy; NHANES III: National Health and Nutrition Examination Survey III; NAFLD: non-alcoholic fatty liver disease; USG: ultrasonography.
Of note, the majority of prevalence studies for lean NAFLD used ultrasound for the detection of liver steatosis, which is a technique that has poor diagnostic accuracy when liver steatosis is below 30%. Therefore, epidemiological studies may have underestimated the true prevalence of NAFLD as an important proportion of individuals with mild liver steatosis may have not been diagnosed as having NAFLD. Indeed, studies using more sensitive methods such as 1H-MRS found a higher prevalence rate of NAFLD (19.3%) [14].
The abovementioned studies evaluated the prevalence of NAFLD; most studies used ultrasonography for evaluation of NAFLD. However, a few studies also evaluated the prevalence of NASH, which is a progressive form of NAFLD. NASH is a less common condition in the general population, with an estimated prevalence in the United States of ~5% [7]. In the NHANES III study, when NAFLD participants with elevated liver enzymes and T2DM or insulin resistance were presumed to have NASH, the prevalence of NASH in non-obese subjects was estimated to be around 0.1% [7]. In an autopsy series of 351 participants, 18.5% of markedly obese subjects and 2.7% of lean subjects were found to have biopsy-proven NASH [21]. In a meta-analysis, among people with lean or non-obese NAFLD, 39% had NASH, 29.2% had significant liver fibrosis (stage ≥ 2), and 3.2% had cirrhosis [17].
Overall, the variability in the prevalence rates of NAFLD and NASH suggests that further studies, using consistent and accurate diagnostic modalities, are needed to provide the true prevalence of lean NAFLD/NASH worldwide. Furthermore, as liver-related and overall mortality in individuals with NAFLD highly depends on the fibrosis stage, a precise estimation of the global prevalence of advanced liver fibrosis in non-obese subjects is needed. Furthermore, longitudinal biopsy-based studies evaluating the progression of each condition of NAFLD spectrum, such as simple steatosis, NASH, and fibrosis, could help evaluate the clinical relevance and outcomes of these conditions in lean/non-obese NAFLD.
3. Mechanisms Underlying Lean and Non-Obese NAFLD
It is important to note that lean and non-obese NAFLD share several pathophysiological mechanisms with obese NAFLD, as illustrated in Figure 1. However, there are unique features underlying lean and non-obese NAFLD (although still not fully understood) that are described as follows.
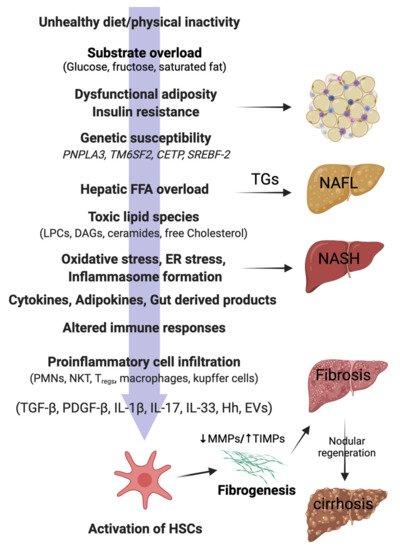
Figure 1. Pathophysiology of NAFLD in lean individuals. In genetically predisposed (PNPLA3, TM6SF2) lean or non-obese individuals, free fatty acids (FFAs), mainly derived from the visceral adipose tissue, are taken up by the liver. Hepatic FFA accumulation can be influenced by the presence of insulin resistance, dysfunctional adiposity, and/or and lifestyle habits (unhealthy diets, physical inactivity). Excess of dietary sugars (glucose, fructose) are converted into FFAs by de novo lipogenesis (DNL). FFAs in the liver mitochondria undergo β-oxidation or are converted back into triglycerides (TGs) for export to the circulation via VLDL. Overwhelming of FFA disposal mechanisms leads to accumulation of TGs as lipid droplets in the hepatocytes (non-alcoholic fatty liver; NAFL). When the FFA pool expands further, cytotoxic lipid species (e.g., LPCs, DAGs, and ceramides) are produced, which ultimately mediate oxidative stress, endoplasmic reticulum (ER) stress, and inflammasome activation. These pathological processes lead to hepatocellular injury, inflammatory cell recruitment, and apoptosis/necroptosis to produce the histological phenotype of non-alcoholic steatohepatitis (NASH). Major modulators of the hepatocellular response to lipotoxic stress may include the gut microbiota products; a variety of cytokines, chemokines, and adipokines; free cholesterol; uric acid; and possibly periodic hypoxia caused by obstructive sleep apnea. Overinduction of inflammatory processes then stimulates hepatic stellate cells (HSC) and activates fibrogenesis. HSC activation is the final common pathway for a diverse group of signals, such as transforming growth factor-beta (TGF-β), platelet-derived growth factor-beta (PDGF-β), interleukins (ILs), hedgehog ligands (Hhs), and extracellular vesicles (EVs). Increased tissue inhibitors of matrix metalloproteinases (TIMPs) cause inhibition of matrix metalloproteinases (MMPs), thereby leading to net gain of fibrosis tissue by the liver. Excessive and disorganized fibrous tissue causes disruption of hepatocellular architecture and nodule formation, leading to cirrhosis of the liver. PNPLA3, patatin-like phospholipase domain containing 3; TM6SF2, Transmembrane 6 superfamily 2; CETP, Cholesteryl ester transfer protein; SREBF-2, sterol regulatory element binding transcription factor 2; LPCs, lysophosphatidylcholines; DAGs, diacyl glycerols; PMNs, polymorphonuclear leucocytes; NKTs, natural killer T cells; Tregs, regulatory T cells.
3.1. Hepatic Lipid Accumulation and Lipotoxicity
The pathophysiology of lean/non-obese NAFLD is similar to obese NAFLD, with the accumulation of free fatty acids (FFAs) in the liver constituting a key process. In this line, the three major sources of hepatic fatty acids are (1) adipose tissue lipolysis, which is the source of around 60% of FFAs present in the circulation (and even higher under insulin resistance conditions) [2]; (2) excessive carbohydrates levels (especially glucose and fructose), which are converted into FFAs by the liver (de novo lipogenesis) and account for about 26% of stored triglycerides; and (3) dietary lipids, which constitute around 15% of triglycerides in the liver [22]. The liver deals with these FFAs’ overload by two mechanisms: (1) FFAs β-oxidation in the mitochondria and (2) re-esterification to triglycerides and subsequent export as very-low density lipoprotein (VLDL) particles. When these disposal mechanisms are overwhelmed, liver stores excess FFAs as triglycerides, leading to hepatic steatosis [2]. Furthermore, excessive β-oxidation leads to formation of reactive oxygen species (ROS) and toxic lipid species, leading to mitochondrial dysfunction, oxidative and endoplasmic reticulum (ER) stress, and inflammasome activation, finally promoting fibrogenesis [23] (Figure 1).
3.2. Insulin Resistance
Insulin resistance is not only a major factor underlying obese NAFLD, but also plays a key role in lean/non-obese NAFLD [13][24]. Insulin resistance contributes to NAFLD directly by increasing de novo lipogenesis (DNL) and indirectly by increasing FFAs’ delivery to the liver via decreased inhibition of lipolysis in the fat depots. In the liver, hyperinsulinemia (secondary to insulin resistance) increases the expression and activity of the transcription factor, sterol response element binding protein 1-c (SREBP-1c), which further increases the expression of all key enzymes required for DNL. These processes lead to accumulation of excess FFAs in the liver [22][23], which further worsens hepatic insulin resistance. This process is mediated by translocation of the PKC-δ isoform from the cytosolic to the membrane compartment, resulting in impairment of hepatic insulin receptor substrate (IRS)-associated phosphatidylinositol 3-kinase (PI3K) activity [25]. The excess of hepatic FFAs is a major driver of NAFLD pathogenesis, secondary to overwhelming of FFA disposal mechanisms and generation of toxic lipid species, such as lysophosphatidylcholines (LPCs), diacylglycerol (DAG), and ceramides. In biopsy-proven NAFLD individuals, circulatory FFAs were significantly elevated in lean, overweight, and obese NAFLD subjects, as compared with healthy controls, with myristic acid (14:0) and palmitoleic acid (16:1) being potential markers for early diagnosis of NAFLD in lean individuals [26]. So, FFAs contribute to peripheral and hepatic insulin resistance, and insulin resistance then leads to further accumulation of hepatic FFAs, thus propagating the vicious cycle.
3.3. Visceral Fat and Metabolic Dysfunction
Distribution of different adipose tissue depots among individuals with similar BMI is a critical factor implicated in metabolic status. Thus, several studies have reported that the contribution of visceral fat to NAFLD is more important than total body fat [13][27][28]. In this line, lean/non-obese individuals with NAFLD have relatively increased amounts of visceral adipose tissue (VAT), as compared with healthy controls [13][27][28]. A recent study noted that a higher visceral-to-subcutaneous fat ratio is associated with increased risk of NAFLD development and advanced fibrosis risk in obese/non-obese NAFLD subjects. VAT is metabolically more active than other adipose tissue depots, and it has been shown to be the source for around 5–10% of FFAs that reach the portal vein blood in lean healthy individuals. Importantly, in people with expanded visceral adiposity, the contribution of VAT to portal vein FFAs’ levels can go up to 50% [29]. Recently, a biopsy-based study including 250 lean, potential living liver donors, the severity of NAFLD was positively correlated with visceral fat accumulation [30].
VAT also constitutes a major source of adipocytokine secretion that contributes to systemic inflammation [31]. Although the specific inflammatory pathways in lean NAFLD pathophysiology are yet to be elucidated, a number of cytokines have been implicated in the development and progression of the disease. Like obesity-related NAFLD, lean individuals with NAFLD present decreased levels of adiponectin, an important hormone with insulin-sensitizing and anti-inflammatory effects [32][33]. Additionally, results from animal models reveal that interleukin 6 (IL-6) overexpression may not only be a consequence but also a central causal factor of NAFLD regardless of the presence of overweight/obesity [34]. Other potential cytokines involved in lean/non-obese NAFLD development are shown in Figure 1.
3.4. Sarcopenia
Low skeletal muscle mass and reduced function, termed as sarcopenia, is another clinical characteristic that may trigger NAFLD in normal-weight individuals. Moreover, several studies have reported the association of sarcopenia with NAFLD complications, such as NASH and liver fibrosis, independently of obesity [35][36][37][38][39][40]. Skeletal muscle mass seems to be lower in lean individuals with NAFLD, as compared with obese people with NAFLD [41][42]. In 762 individuals with biopsy-proven NAFLD, skeletal muscle mass was significantly lower in non-obese patients [42]. From Japan, sarcopenic obesity was significantly associated with non-obese NAFLD, and the association persisted even after adjusting for metabolic confounders (OR, 2.367; 95% CI, 1.317–4.254, p = 0.004) [43]. The same authors demonstrated a significant association of osteosarcopenic obesity with non-obese NAFLD in females [44]. However, the direction of cause-and-effect relationship is unknown.
Insulin resistance contributes to loss of lean body mass (sarcopenia) by the activation of ubiquitin-proteasome proteolytic pathway (UPP) in skeletal muscle. Insulin resistance reduces the activity of PI3K, which, in turn, reduces the levels of phosphorylated Akt. The decrease in phosphorylated Akt levels induces the expression and activity of E3 ubiquitin-conjugating enzymes, thereby activating UPP [45].
The muscle is the primary organ responsible for insulin-mediated glucose disposal; hence, a decrease in muscle mass may cause impairment in glucose metabolism [46]. Sarcopenia also decreases the tolerance to exercise, further decreasing energy expenditure and promoting weight gain and insulin resistance [47]. Myokines may play a role in the development in NAFLD, especially in lean and non-obese individuals. In this line, exercise stimulates the expression of peroxisome proliferator-activated receptor γ (PPAR-γ) coactivator-1α (PGC-1α), which is accompanied by greater fibronectin type III domain-containing 5 transmembrane receptor (FNCD5) membrane expression. FNDC5 is cleaved, releasing irisin, an exercise-inducible myokine that causes browning of white adipose tissue, increasing energy expenditure due to heat loss and weight loss [48]. In the liver, irisin has direct anti-steatogenic effects through activation of PPAR-α and upregulation of fibroblast growth factor 21 (FGF21) [49][50]. Several studies [51][52], but not all [53], have shown a negative correlation between irisin and hepatic steatosis severity.
Myostatin is another myokine that is downregulated by exercise [54]. Myostatin is an inhibitor of muscle growth since it promotes proteolysis and inhibits muscle regeneration and function [55]. Myostatin also has metabolic actions, promoting adipose tissue expansion through direct effects on the adipose tissue and indirectly through downregulation of irisin [56][57]. High myostatin concentration is associated with insulin resistance, which, in turn, is associated with muscle atrophy, decreased exercise capacity, and metabolic defects [58]. Myostatin has been shown to negatively correlate with lean mass in healthy adults [59]. Lastly, myostatin has fibrogenic properties through direct action on hepatic stellate cells [60]. Interestingly, myostatin levels correlate with liver steatosis in lean subjects [41].
On the other hand, chronic inflammation due to abnormal cytokine production in NAFLD and ROS overload have also deleterious effects on skeletal muscle, especially in lean/non-obese NAFLD. Chronic exposure to IL-6 leads to increased muscle catabolism and atrophy [61]. However, further studies are needed to establish this observation. Elevated levels of tumor necrosis factor alpha (TNF-α) induce ceramide accumulation that has been observed to contribute to muscle–cell atrophy [62]. C-reactive protein levels (CRP) correlate with the loss of total appendicular skeletal muscle [63]. Additionally, there is a well-known interplay between insulin resistance and sarcopenia. In this context, the complex crosstalk between the liver and the muscle results in a vicious circle with equally harmful consequences for both organs.
Finally, it is important to highlight that visceral adiposity and sarcopenia seem to act synergistically in the pathogenesis and progression of NAFLD. Thus, in patients with NAFLD, a decrease in muscle mass and increase in VAT are associated with worsening of steatosis and progression of liver fibrosis [64]. In this line, the assessment of body composition through different methods (e.g., bioelectrical impedance analysis (BIA), ultrasound, computed tomography (CT), magnetic resonance imaging (MRI)) could become a useful tool to detect high-risk patients. Thus, in a longitudinal cohort including almost 10,000 subjects without NAFLD (mean BMI of 22.3 kg/m2), body composition measurement by BIA showed that, at baseline, the lowest and middle weight-adjusted skeletal muscle index (SMI) tertiles and an increased fat percentage were associated with incident NAFLD in individuals with normal weight, while the SMI increase between examinations was an independent protective factor against NAFLD [65].
3.5. Genetic Predisposition
Variation in the patatin-like phospholipase domain-containing 3 (PNPLA3) gene is strongly linked to differences in liver fat content and susceptibility to NAFLD in lean and non-obese individuals [14][32][66][67]. PNPLA3 is expressed in both adipocytes and hepatocytes and has acyl hydrolase activity, which leads to the hydrolysis of monoacylglycerol, diacylglycerol, and triacylglycerol [68]. The rs738409 polymorphism is associated with the loss of protein’s hydrolyzing function, thereby resulting in liver fat accumulation and insulin resistance, irrespective of body weight [69]. Indeed, a meta-analysis reported that the rs738409 polymorphism is more prevalent in non-obese/lean NAFLD patients than in obese NAFLD and non-obese controls [19].
On the other hand, genome-wide association studies found a link between NAFLD and TM6SF2 [70][71]. Thus, the TM6SF2 rs58542926 C > T (E167K) allele influences hepatic fibrosis irrespective of confounding risk factors such as obesity, T2DM, and age [72]. Notably, this variant has been associated with lower BMI and peripheral fat in individuals with NAFLD in phenome-wide association studies [73] Moreover, recent studies on biopsy-proven lean/non-obese NAFLD individuals reported a direct relationship between the TM6SF2 T allele and lean/non-obese NAFLD [74][75].
Cholesteryl ester transfer protein (CETP) gene polymorphisms are associated with an increased risk of lean NAFLD [76]. Two single-nucleotide polymorphisms in CETP (rs12447924 and rs12597002) have been associated with non-obese NAFLD. The probability of lean NAFLD was above 30% in lean homozygotes, 10–15% in lean heterozygotes, and 3–5% in lean wild types, while the probability of NAFLD in obese patients was over 30% in all genotypes [76]. Furthermore, a polymorphism in the sterol regulatory element-binding factor-2 (SREBF-2) gene was found to be associated with lean and non-obese NAFLD [77]. In addition, it was noted that SREBF-2 polymorphism has a significant impact on lipid and glucose metabolism and liver histology in biopsy-proven NAFLD patients and predisposes healthy individuals to develop non-obese NAFLD [77]. It was found that insufficiency of phosphatidylethanolamine N-methyltransferase (PEMT) increased the risk of NASH in lean individuals [78]. Another genetic factor that could play a role in lean or non-obese NAFLD is the rs368234815 TT polymorphism in the interferon lambda 4 (IFNL4) gene [79]. Recently, the gene of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), encoded by PPARGC1A, was found to be a susceptibility candidate gene for NAFLD. Moreover, PPARGC1A rs8192678 A allele was also found to be a risk factor for non-obese NASH (OR, 22.00; 95% CI, 1.54–314.29; p = 0.021) [80]. However, further large studies are required to confirm the role of CETP, SREBF-2, PEMT, IFNL4, and PPARGC1A gene polymorphisms in the susceptibility and pathogenesis of NAFLD in lean or non-obese individuals.
It is important to highlight that gene–environment interactions (e.g., diet, physical activity, metabolic comorbidities, or gut microbiota) seem to be crucial to modulate gene polymorphism-mediated liver damage in lean/non-obese NAFLD. The low prevalence of NAFLD in lean subjects without altered metabolic profile carrying some of these mutations makes it unlikely that the presence of a risk variant itself determines the development and progression of NAFLD [81]. Thus, on a predisposed genetic background, environmental factors could trigger the disease and related complications. Intriguingly, a recent cohort study of 1339 biopsy-proven NAFLD Caucasian patients (195 lean, BMI < 25 kg/m2) showed that NAFLD development and progression in lean individuals were independent of their PNPLA3 genotype [82].
In light of the many studies discussed above, further research is needed to assess the role of genetic determinants in NAFLD pathophysiology and the impact of their interplay with other risk factors.
3.6. Gut Microbiota Dysbiosis
Gut microbial dysbiosis is characterized by an increase in pathogenic bacteria and a decrease in number (abundance) and diversity (richness) of beneficial bacteria [83]. The mechanisms by which intestinal dysbiosis contributes to NAFLD include dysbiosis-induced gut permeability, endotoxemia, endogenous ethanol production, increased energy harvest from food, and alterations in choline and bile acid metabolism [83].
In this research line, there is growing evidence that individuals with lean NAFLD have a distinct gut microbiota profile with respect to obese individuals with NAFLD (Table 2). Thus, in a study conducted in a Chinese population, non-obese patients with NAFLD demonstrated reductions in Firmicutes including Lachnospiraceae, Ruminococcaceae, and Lactobacillacea, and an increase in lipopolysaccharide-producing Gram-negative bacteria [84]. Additionally, in a pilot study with biopsy-proven patients, lean NASH individuals had a lower abundance of Faecalibacterium, Ruminococcus, and Lactobacillus compared with non-lean individuals with NASH [85]. In another study, a decrease in Desulfovibrionaceae was found to be associated with lean NAFLD, compared to obese NAFLD individuals [86]. Additionally, in another study with biopsy-proven NAFLD, along with an enrichment of Veillonellaceae and a depletion of Ruminococcaceae with the worsening of liver fibrosis in individuals with non-obese NAFLD, there were increased levels of total bile acids and propionate [87]. Finally, a study conducted on a Japanese cohort of patients showed a significant decrease in the abundance of Eubacterium in non-obese subjects with NAFLD [88]. In another biopsy-proven cohort of 538 Caucasians patients with NAFLD, lean NAFLD subjects had higher total bile acid and FGF19 levels compared to those with non-lean NAFLD and also showed decreased levels of butyric acid [74]. Further studies are required to clearly delineate whether the changes observed in gut microbiota composition of lean individuals are either the cause or consequence of NAFLD.
Table 2. Clinical studies of gut microbiota in individuals with lean NAFLD.
| Study, Year | Subjects | Diagnosis of NAFLD | Lean or Non-Obese | Decreased Abundance Associated with NAFLD | Increased Abundance Associated with NAFLD |
|---|---|---|---|---|---|
| Wang et al., 2016 [84] | 126 non-obese subjects | USG | Non-obese | Lachnospiraceae Ruminococcaceae Lactobacillaceae |
LPS-producing Gram negative bacteria |
| Duarte et al., 2018 [85] | 13 NASH; 10 controls |
Biopsy | Lean | Faecalibacterium Ruminococcus lactobacillus |
- |
| Yun et al., 2019 [86] | 268 health check-up examinees | USG | Non-obese | Desulfovibrionaceae | - |
| Lee et al., 2020 [87] | 171 Asians | Biopsy | Non-obese | Ruminococcaceae | Veillonellaceae |
| Iwaki et al., 2021 [88] | 51 non-obese NAFLD; 51 obese NAFLD; 87 controls | Biopsy | Non-obese | Eubacterium | - |
LPS, lipopolysaccharide; NAFLD, non-alcoholic fatty liver disease; USG, ultrasonography.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9101346
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20.
- Kuchay, M.S.; Choudhary, N.S.; Mishra, S.K. Pathophysiological Mechanisms Underlying MAFLD. Diabetes Metab. Syndr. 2020, 14, 1875–1887.
- Weir, C.B.; Jan, A. BMI Classification Percentile and Cut Off Points. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- WHO Expert Consultation. Appropriate Body-Mass Index for Asian Populations and Its Implications for Policy and Intervention Strategies. Lancet Lond. Engl. 2004, 363, 157–163.
- Vilarinho, S.; Ajmera, V.; Zheng, M.; Loomba, R. Emerging Role of Genomic Analysis in Clinical Evaluation of Lean Individuals with NAFLD. Hepatol. Baltim. Md. 2021, 74, 2241–2250.
- Younes, R.; Bugianesi, E. NASH in Lean Individuals. Semin. Liver Dis. 2019, 39, 86–95.
- Younossi, Z.M.; Stepanova, M.; Negro, F.; Hallaji, S.; Younossi, Y.; Lam, B.; Srishord, M. Nonalcoholic Fatty Liver Disease in Lean Individuals in the United States. Medicine 2012, 91, 319–327.
- Zou, B.; Yeo, Y.H.; Nguyen, V.H.; Cheung, R.; Ingelsson, E.; Nguyen, M.H. Prevalence, Characteristics and Mortality Outcomes of Obese, Nonobese and Lean NAFLD in the United States, 1999–2016. J. Intern. Med. 2020, 288, 139–151.
- Sinn, D.H.; Gwak, G.-Y.; Park, H.N.; Kim, J.E.; Min, Y.W.; Kim, K.M.; Kim, Y.J.; Choi, M.S.; Lee, J.H.; Koh, K.C.; et al. Ultrasonographically Detected Non-Alcoholic Fatty Liver Disease Is an Independent Predictor for Identifying Patients with Insulin Resistance in Non-Obese, Non-Diabetic Middle-Aged Asian Adults. Am. J. Gastroenterol. 2012, 107, 561–567.
- Kwon, Y.-M.; Oh, S.-W.; Hwang, S.; Lee, C.; Kwon, H.; Chung, G.E. Association of Nonalcoholic Fatty Liver Disease with Components of Metabolic Syndrome According to Body Mass Index in Korean Adults. Am. J. Gastroenterol. 2012, 107, 1852–1858.
- Kim, Y.; Han, E.; Lee, J.S.; Lee, H.W.; Kim, B.K.; Kim, M.K.; Kim, H.S.; Park, J.Y.; Kim, D.Y.; Ahn, S.H.; et al. Cardiovascular Risk Is Elevated in Lean Subjects with Nonalcoholic Fatty Liver Disease. Gut Liver 2021. .
- Xu, C.; Yu, C.; Ma, H.; Xu, L.; Miao, M.; Li, Y. Prevalence and Risk Factors for the Development of Nonalcoholic Fatty Liver Disease in a Nonobese Chinese Population: The Zhejiang Zhenhai Study. Am. J. Gastroenterol. 2013, 108, 1299–1304.
- Feng, R.-N.; Du, S.-S.; Wang, C.; Li, Y.-C.; Liu, L.-Y.; Guo, F.-C.; Sun, C.-H. Lean-non-alcoholic fatty liver disease increases risk for metabolic disorders in a normal weight Chinese population. World J. Gastroenterol. 2014, 20, 17932–17940.
- Wei, J.L.; Leung, J.C.-F.; Loong, T.C.-W.; Wong, G.L.-H.; Yeung, D.K.-W.; Chan, R.; Chan, H.L.-Y.; Chim, A.M.-L.; Woo, J.; Chu, W.; et al. Prevalence and Severity of Nonalcoholic Fatty Liver Disease in Non-Obese Patients: A Population Study Using Proton-Magnetic Resonance Spectroscopy. Am. J. Gastroenterol. 2015, 110, 1306–1314.
- Nishioji, K.; Sumida, Y.; Kamaguchi, M.; Mochizuki, N.; Kobayashi, M.; Nishimura, T.; Yamaguchi, K.; Itoh, Y. Prevalence of and risk factors for non-alcoholic fatty liver disease in a non-obese Japanese population, 2011–2012. J. Gastroenterol. 2014, 50, 95–108.
- Choudhary, N.S.; Saraf, N.; Saigal, S.; Duseja, A.; Gautam, D.; Rastogi, A.; Bhangui, P.; Thiagrajan, S.; Soin, A.S. Nonalcoholic Fatty Liver in Lean Individuals: Clinicobiochemical Correlates of Histopathology in 157 Liver Biopsies from Healthy Liver Donors. J. Clin. Exp. Hepatol. 2021, 11, 544–549.
- Ye, Q.; Zou, B.; Yeo, Y.H.; Li, J.; Huang, D.Q.; Wu, Y.; Yang, H.; Liu, C.; Kam, L.Y.; Tan, X.X.E.; et al. Global prevalence, incidence, and outcomes of non-obese or lean non-alcoholic fatty liver disease: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 739–752.
- Shi, Y.; Wang, Q.; Sun, Y.; Zhao, X.; Kong, Y.; Ou, X.; Jia, J.; Wu, S.; You, H. The Prevalence of Lean/Nonobese Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. J. Clin. Gastroenterol. 2020, 54, 378–387.
- Zou, Z.Y.; Wong, V.W.-S.; Fan, J.G. Epidemiology of Nonalcoholic Fatty Liver Disease in Non-Obese Populations: Meta-Analytic Assessment of Its Prevalence, Genetic, Metabolic, and Histological Profiles. J. Dig. Dis. 2020, 21, 372–384.
- Lu, F.-B.; Zheng, K.I.; Rios, R.S.; Targher, G.; Byrne, C.D.; Zheng, M.-H. Global Epidemiology of Lean Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. J. Gastroenterol. Hepatol. 2020, 35, 2041–2050.
- Wanless, I.R.; Lentz, J.S. Fatty Liver Hepatitis (Steatohepatitis) and Obesity: An Autopsy Study with Analysis of Risk Factors. Hepatol. Baltim. Md. 1990, 12, 1106–1110.
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351.
- Zhang, N.-P.; Liu, X.-J.; Xie, L.; Shen, X.-Z.; Wu, J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Investig. 2019, 99, 749–763.
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642.
- Lam, T.K.T.; Carpentier, A.; Lewis, G.F.; Van De Werve, G.; Fantus, I.G.; Giacca, A. Mechanisms of the free fatty acid-induced increase in hepatic glucose production. Am. J. Physiol. Metab. 2003, 284, E863–E873.
- Feng, R.; Luo, C.; Li, C.; Du, S.; Okekunle, A.P.; Li, Y.; Chen, Y.; Zi, T.; Niu, Y. Free Fatty Acids Profile among Lean, Overweight and Obese Non-Alcoholic Fatty Liver Disease Patients: A Case—Control Study. Lipids Health Dis. 2017, 16, 165.
- Ha, Y.; Seo, N.; Shim, J.H.; Kim, S.Y.; Park, J.-A.; Han, S.; Kim, K.W.; Yu, E.; Kim, K.M.; Lim, Y.-S.; et al. Intimate association of visceral obesity with non-alcoholic fatty liver disease in healthy Asians: A case-control study. J. Gastroenterol. Hepatol. 2015, 30, 1666–1672.
- Petta, S.; Amato, M.C.; Di Marco, V.; Cammà, C.; Pizzolanti, G.; Barcellona, M.R.; Cabibi, D.; Galluzzo, A.; Sinagra, D.; Giordano, C.; et al. Visceral Adiposity Index Is Associated with Significant Fibrosis in Patients with Non-Alcoholic Fatty Liver Disease. Aliment. Pharmacol. Ther. 2012, 35, 238–247.
- Nielsen, S.; Guo, Z.; Johnson, C.M.; Hensrud, D.D.; Jensen, M.D. Splanchnic lipolysis in human obesity. J. Clin. Investig. 2004, 113, 1582–1588.
- Lee, S.; Kim, K.W.; Lee, J.; Park, T.; Khang, S.; Jeong, H.; Song, G.; Lee, S. Visceral Adiposity as a Risk Factor for Lean Nonalcoholic Fatty Liver Disease in Potential Living Liver Donors. J. Gastroenterol. Hepatol. 2021. .
- Fontana, L.; Eagon, J.C.; Trujillo, M.E.; Scherer, P.E.; Klein, S. Visceral Fat Adipokine Secretion Is Associated With Systemic Inflammation in Obese Humans. Diabetes 2007, 56, 1010–1013.
- Feldman, A.; Eder, S.K.; Felder, T.K.; Kedenko, L.; Paulweber, B.; Stadlmayr, A.; Huber-Schönauer, U.; Niederseer, D.; Stickel, F.; Auer, S.; et al. Clinical and Metabolic Characterization of Lean Caucasian Subjects With Non-Alcoholic Fatty Liver. Am. J. Gastroenterol. 2017, 112, 102–110.
- Woodward, L.; Akoumianakis, I.; Antoniades, C. Unravelling the adiponectin paradox: Novel roles of adiponectin in the regulation of cardiovascular disease. Br. J. Pharmacol. 2016, 174, 4007–4020.
- Singh, M.K.; Jayarajan, R.; Varshney, S.; Upadrasta, S.; Singh, A.; Yadav, R.; Scaria, V.; Sengupta, S.; Shanmugam, D.; Sivasubbu, S.; et al. Chronic systemic exposure to IL6 leads to deregulation of glycolysis and fat accumulation in the zebrafish liver. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2021, 1866, 158905.
- Lee, Y.; Kim, S.U.; Song, K.; Park, J.Y.; Kim, D.Y.; Ahn, S.H.; Lee, B.-W.; Kang, E.S.; Cha, B.-S.; Han, K.-H. Sarcopenia Is Associated with Significant Liver Fibrosis Independently of Obesity and Insulin Resistance in Nonalcoholic Fatty Liver Disease: Nationwide Surveys (KNHANES 2008–2011). Hepatol. Baltim. Md. 2016, 63, 776–786.
- Koo, B.K.; Kim, D.; Joo, S.K.; Kim, J.H.; Chang, M.S.; Kim, B.G.; Lee, K.L.; Kim, W. Sarcopenia Is an Independent Risk Factor for Non-Alcoholic Steatohepatitis and Significant Fibrosis. J. Hepatol. 2017, 66, 123–131.
- Petta, S.; Ciminnisi, S.; Di Marco, V.; Cabibi, D.; Cammà, C.; Licata, A.; Marchesini, G.; Craxì, A. Sarcopenia is associated with severe liver fibrosis in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2017, 45, 510–518.
- Yu, R.; Shi, Q.; Liu, L.; Chen, L. Relationship of sarcopenia with steatohepatitis and advanced liver fibrosis in non-alcoholic fatty liver disease: A meta-analysis. BMC Gastroenterol. 2018, 18, 51.
- Cai, C.; Song, X.; Chen, Y.; Chen, X.; Yu, C. Relationship between Relative Skeletal Muscle Mass and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Hepatol. Int. 2020, 14, 115–126.
- Kuchay, M.S.; Choudhary, N.S.; Gagneja, S.; Mathew, A.; Bano, T.; Kaur, P.; Bahadur, B.; Singh, M.K.; Gill, H.K.; Wasir, J.S.; et al. Low skeletal muscle mass is associated with liver fibrosis in individuals with type 2 diabetes and NAFLD. J. Gastroenterol. Hepatol. 2021. .
- Shida, T.; Oshida, N.; Suzuki, H.; Okada, K.; Watahiki, T.; Oh, S.; Kim, T.; Isobe, T.; Okamoto, Y.; Ariizumi, S.-I.; et al. Clinical and Anthropometric Characteristics of Non-Obese Non-Alcoholic Fatty Liver Disease Subjects in Japan. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2020, 50, 1032–1046.
- Tobari, M.; Hashimoto, E.; Taniai, M.; Ikarashi, Y.; Kodama, K.; Kogiso, T.; Tokushige, K.; Takayoshi, N.; Hashimoto, N. Characteristics of Non-Alcoholic Steatohepatitis among Lean Patients in Japan: Not Uncommon and Not Always Benign. J. Gastroenterol. Hepatol. 2019, 34, 1404–1410.
- Kashiwagi, K.; Takayama, M.; Fukuhara, K.; Shimizu-Hirota, R.; Chu, P.-S.; Nakamoto, N.; Inoue, N.; Iwao, Y.; Kanai, T. A significant association of non-obese non-alcoholic fatty liver disease with sarcopenic obesity. Clin. Nutr. ESPEN 2020, 38, 86–93.
- Kashiwagi, K.; Takayama, M.; Ichikawa, H.; Takaishi, H.; Iwao, Y.; Kanai, T. A significant association of non-obese non-alcoholic fatty liver disease with osteosarcopenic obesity in females 50 years and older. Clin. Nutr. ESPEN 2021, 42, 166–172.
- Wang, X.; Hu, Z.; Hu, J.; Du, J.; Mitch, W.E. Insulin Resistance Accelerates Muscle Protein Degradation: Activation of the Ubiquitin-Proteasome Pathway by Defects in Muscle Cell Signaling. Endocrinology 2006, 147, 4160–4168.
- DeFronzo, R.A. From the Triumvirate to the Ominous Octet: A New Paradigm for the Treatment of Type 2 Diabetes Mellitus. Diabetes 2009, 58, 773–795.
- Kim, T.N.; Yang, S.J.; Yoo, H.J.; Lim, K.I.; Kang, H.J.; Song, W.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; et al. Prevalence of Sarcopenia and Sarcopenic Obesity in Korean Adults: The Korean Sarcopenic Obesity Study. Int. J. Obes. 2009, 33, 885–892.
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-Dependent Myokine That Drives Brown-Fat-like Development of White Fat and Thermogenesis. Nature 2012, 481, 463–468.
- Park, M.-J.; Kim, D.-I.; Choi, J.-H.; Heo, Y.-R.; Park, S.-H. New role of irisin in hepatocytes: The protective effect of hepatic steatosis in vitro. Cell. Signal. 2015, 27, 1831–1839.
- Bhanji, R.A.; Narayanan, P.; Allen, A.M.; Malhi, H.; Watt, K.D. Sarcopenia in Hiding: The Risk and Consequence of Underestimating Muscle Dysfunction in Nonalcoholic Steatohepatitis. Hepatol. Baltim. Md. 2017, 66, 2055–2065.
- Zhang, H.-J.; Zhang, X.-F.; Ma, Z.-M.; Pan, L.-L.; Chen, Z.; Han, H.-W.; Han, C.-K.; Zhuang, X.-J.; Lu, Y.; Li, X.-J.; et al. Irisin is inversely associated with intrahepatic triglyceride contents in obese adults. J. Hepatol. 2013, 59, 557–562.
- Metwally, M.; Bayoumi, A.; Romero-Gomez, M.; Thabet, K.; John, M.; Adams, L.A.; Huo, X.; Aller, R.; García-Monzón, C.; Teresa Arias-Loste, M.; et al. A Polymorphism in the Irisin-Encoding Gene (FNDC5) Associates with Hepatic Steatosis by Differential MiRNA Binding to the 3′UTR. J. Hepatol. 2019, 70, 494–500.
- Choi, E.S.; Kim, M.K.; Song, M.K.; Kim, J.M.; Kim, E.S.; Chung, W.J.; Park, K.S.; Cho, K.B.; Hwang, J.S.; Jang, B.K. Association between Serum Irisin Levels and Non-Alcoholic Fatty Liver Disease in Health Screen Examinees. PLoS ONE 2014, 9, e110680.
- Huh, J.Y. The role of exercise-induced myokines in regulating metabolism. Arch. Pharm. Res. 2017, 41, 14–29.
- Han, H.; Zhou, X.; Mitch, W.E.; Goldberg, A.L. Myostatin/activin pathway antagonism: Molecular basis and therapeutic potential. Int. J. Biochem. Cell Biol. 2013, 45, 2333–2347.
- Li, F.; Li, Y.; Duan, Y.; Hu, C.-A.A.; Tang, Y.; Yin, Y. Myokines and Adipokines: Involvement in the Crosstalk between Skeletal Muscle and Adipose Tissue. Cytokine Growth Factor Rev. 2017, 33, 73–82.
- Konopka, A.R.; Wolff, C.A.; Suer, M.K.; Harber, M.P. Relationship between Intermuscular Adipose Tissue Infiltration and Myostatin before and after Aerobic Exercise Training. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R461–R468.
- Nishikawa, H.; Enomoto, H.; Ishii, A.; Iwata, Y.; Miyamoto, Y.; Ishii, N.; Yuri, Y.; Hasegawa, K.; Nakano, C.; Nishimura, T.; et al. Elevated Serum Myostatin Level Is Associated with Worse Survival in Patients with Liver Cirrhosis. J. Cachexia Sarcopenia Muscle 2017, 8, 915–925.
- Gonzalez-Cadavid, N.F.; Taylor, W.E.; Yarasheski, K.; Sinha-Hikim, I.; Ma, K.; Ezzat, S.; Shen, R.; Lalani, R.; Asa, S.; Mamita, M.; et al. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc. Natl. Acad. Sci. USA 1998, 95, 14938–14943.
- Delogu, W.; Caligiuri, A.; Provenzano, A.; Rosso, C.; Bugianesi, E.; Coratti, A.; Macias-Barragan, J.; Galastri, S.; Di Maira, G.; Marra, F. Myostatin regulates the fibrogenic phenotype of hepatic stellate cells via c-jun N-terminal kinase activation. Dig. Liver Dis. 2019, 51, 1400–1408.
- Haddad, F.; Zaldivar, F.; Cooper, D.M.; Adams, G.R. IL-6-induced skeletal muscle atrophy. J. Appl. Physiol. 2005, 98, 911–917.
- De Larichaudy, J.; Zufferli, A.; Serra, F.; Isidori, A.M.; Naro, F.; Dessalle, K.; Desgeorges, M.; Piraud, M.; Cheillan, D.; Vidal, H.; et al. TNF-α- and tumor-induced skeletal muscle atrophy involves sphingolipid metabolism. Skelet. Muscle 2012, 2, 2.
- Alemán, H.; Esparza, J.; Ramirez, F.A.; Astiazaran, H.; Payette, H. Longitudinal Evidence on the Association between Interleukin-6 and C-Reactive Protein with the Loss of Total Appendicular Skeletal Muscle in Free-Living Older Men and Women. Age Ageing 2011, 40, 469–475.
- Shida, T.; Oshida, N.; Oh, S.; Okada, K.; Shoda, J. Progressive reduction in skeletal muscle mass to visceral fat area ratio is associated with a worsening of the hepatic conditions of non-alcoholic fatty liver disease. Diabetes Metab. Syndr. Obesity Targets Ther. 2019, 12, 495–503.
- Kim, H.Y.; Baik, S.J.; Lee, H.A.; Lee, B.K.; Lee, H.S.; Kim, T.H.; Yoo, K. Relative Fat Mass at Baseline and Its Early Change May Be a Predictor of Incident Nonalcoholic Fatty Liver Disease. Sci. Rep. 2020, 10, 17491.
- Honda, Y.; Yoneda, M.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Mawatari, H.; Fujita, K.; Hyogo, H.; Ueno, T.; et al. Characteristics of Non-Obese Non-Alcoholic Fatty Liver Disease: Effect of Genetic and Environmental Factors. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2016, 46, 1011–1018.
- Niriella, M.A.; Kasturiratne, A.; Pathmeswaran, A.; De Silva, S.T.; Perera, K.R.; Subasinghe, S.K.C.E.; Kodisinghe, S.K.; Piyaratna, T.A.C.L.; Vithiya, K.; Dassanayaka, A.S.; et al. Lean Non-Alcoholic Fatty Liver Disease (Lean NAFLD): Characteristics, Metabolic Outcomes and Risk Factors from a 7-Year Prospective, Community Cohort Study from Sri Lanka. Hepatol. Int. 2019, 13, 314–322.
- Huang, Y.; Cohen, J.C.; Hobbs, H.H. Expression and Characterization of a PNPLA3 Protein Isoform (I148M) Associated with Nonalcoholic Fatty Liver Disease. J. Biol. Chem. 2011, 286, 37085–37093.
- Oniki, K.; Saruwatari, J.; Izuka, T.; Kajiwara, A.; Morita, K.; Sakata, M.; Otake, K.; Ogata, Y.; Nakagawa, K. Influence of the PNPLA3 Rs738409 Polymorphism on Non-Alcoholic Fatty Liver Disease and Renal Function among Normal Weight Subjects. PLoS ONE 2015, 10, e0132640.
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 Superfamily Member 2 Gene Variant Disentangles Nonalcoholic Steatohepatitis from Cardiovascular Disease. Hepatol. Baltim. Md. 2015, 61, 506–514.
- Anstee, Q.M.; Darlay, R.; Cockell, S.; Meroni, M.; Govaere, O.; Tiniakos, D.; Burt, A.D.; Bedossa, P.; Palmer, J.; Liu, Y.-L.; et al. Genome-Wide Association Study of Non-Alcoholic Fatty Liver and Steatohepatitis in a Histologically Characterised Cohort. J. Hepatol. 2020, 73, 505–515.
- Liu, Y.-L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.S.; Allison, M.E.D.; Alexander, G.J.; Piguet, A.-C.; Anty, R.; et al. TM6SF2 Rs58542926 Influences Hepatic Fibrosis Progression in Patients with Non-Alcoholic Fatty Liver Disease. Nat. Commun. 2014, 5, 4309.
- Chen, V.L.; Chen, Y.; Du, X.; Handelman, S.K.; Speliotes, E.K. Genetic Variants That Associate with Cirrhosis Have Pleiotropic Effects on Human Traits. Liver Int. Off. J. Int. Assoc. Study Liver 2020, 40, 405–415.
- Chen, F.; Esmaili, S.; Rogers, G.B.; Bugianesi, E.; Petta, S.; Marchesini, G.; Bayoumi, A.; Metwally, M.; Azardaryany, M.K.; Coulter, S.; et al. Lean NAFLD: A Distinct Entity Shaped by Differential Metabolic Adaptation. Hepatol. Baltim. Md. 2020, 71, 1213–1227.
- Wang, Q.; You, H.; Ou, X.; Zhao, X.; Sun, Y.; Wang, M.; Wang, P.; Wang, Y.; Duan, W.; Wang, X.; et al. Non-Obese Histologically Confirmed NASH Patients with Abnormal Liver Biochemistry Have More Advanced Fibrosis. Hepatol. Int. 2019, 13, 766–776.
- Adams, L.A.; Marsh, J.A.; Ayonrinde, O.T.; Olynyk, J.K.; Ang, W.Q.; Beilin, L.J.; Mori, T.; Palmer, L.J.; Oddy, W.W.; Lye, S.J.; et al. Cholesteryl Ester Transfer Protein Gene Polymorphisms Increase the Risk of Fatty Liver in Females Independent of Adiposity. J. Gastroenterol. Hepatol. 2012, 27, 1520–1527.
- Musso, G.; Cassader, M.; Bo, S.; De Michieli, F.; Gambino, R. Sterol Regulatory Element-Binding Factor 2 (SREBF-2) Predicts 7-Year NAFLD Incidence and Severity of Liver Disease and Lipoprotein and Glucose Dysmetabolism. Diabetes 2013, 62, 1109–1120.
- Nakatsuka, A.; Matsuyama, M.; Yamaguchi, S.; Katayama, A.; Eguchi, J.; Murakami, K.; Teshigawara, S.; Ogawa, D.; Wada, N.; Yasunaka, T.; et al. Insufficiency of Phosphatidylethanolamine N-Methyltransferase Is Risk for Lean Non-Alcoholic Steatohepatitis. Sci. Rep. 2016, 6, 21721.
- Petta, S.; Valenti, L.; Tuttolomondo, A.; Dongiovanni, P.; Pipitone, R.M.; Cammà, C.; Cabibi, D.; Di Marco, V.; Fracanzani, A.L.; Badiali, S.; et al. Interferon lambda 4 rs368234815 TT>δG variant is associated with liver damage in patients with nonalcoholic fatty liver disease. Hepatology 2017, 66, 1885–1893.
- Zhang, R.-N.; Shen, F.; Pan, Q.; Cao, H.-X.; Chen, G.-Y.; Fan, J.-G. PPARGC1A Rs8192678 G>A Polymorphism Affects the Severity of Hepatic Histological Features and Nonalcoholic Steatohepatitis in Patients with Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2021, 27, 3863–3876.
- Mann, J.P.; Anstee, Q.M. NAFLD: PNPLA3 and Obesity: A Synergistic Relationship in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 506–507.
- Vos, B.; Moreno, C.; Nagy, N.; Féry, F.; Cnop, M.; Vereerstraeten, P.; Devière, J.; Adler, M. Lean Non-Alcoholic Fatty Liver Disease (Lean-NAFLD): A Major Cause of Cryptogenic Liver Disease. Acta Gastro-Enterol. Belg. 2011, 74, 389–394.
- Safari, Z.; Gérard, P. The Links between the Gut Microbiome and Non-Alcoholic Fatty Liver Disease (NAFLD). Cell. Mol. Life Sci. CMLS 2019, 76, 1541–1558.
- Wang, B.; Jiang, X.; Cao, M.; Ge, J.; Bao, Q.; Tang, L.; Chen, Y.; Li, L. Altered Fecal Microbiota Correlates with Liver Biochemistry in Nonobese Patients with Non-Alcoholic Fatty Liver Disease. Sci. Rep. 2016, 6, 32002.
- Duarte, S.M.B.; Stefano, J.T.; Miele, L.; Ponziani, F.R.; Souza-Basqueira, M.; Okada, L.S.R.R.; de Barros Costa, F.G.; Toda, K.; Mazo, D.F.C.; Sabino, E.C.; et al. Gut Microbiome Composition in Lean Patients with NASH Is Associated with Liver Damage Independent of Caloric Intake: A Prospective Pilot Study. Nutr. Metab. Cardiovasc. Dis. NMCD 2018, 28, 369–384.
- Yun, Y.; Kim, H.-N.; Lee, E.-J.; Ryu, S.; Chang, Y.; Shin, H.; Kim, H.-L.; Kim, T.H.; Yoo, K.; Kim, H.Y. Fecal and Blood Microbiota Profiles and Presence of Nonalcoholic Fatty Liver Disease in Obese versus Lean Subjects. PLoS ONE 2019, 14, e0213692.
- Lee, G.; You, H.J.; Bajaj, J.S.; Joo, S.K.; Yu, J.; Park, S.; Kang, H.; Park, J.H.; Kim, J.H.; Lee, D.H.; et al. Distinct Signatures of Gut Microbiome and Metabolites Associated with Significant Fibrosis in Non-Obese NAFLD. Nat. Commun. 2020, 11, 4982.
- Iwaki, M.; Kessoku, T.; Ozaki, A.; Kasai, Y.; Kobayashi, T.; Nogami, A.; Honda, Y.; Ogawa, Y.; Imajo, K.; Yoneda, M.; et al. Gut Microbiota Composition Associated with Hepatic Fibrosis in Non-Obese Patients with Non-Alcoholic Fatty Liver Disease. J. Gastroenterol. Hepatol. 2021, 36, 2275–2284.
This entry is offline, you can click here to edit this entry!