Melanoma is the least common form of skin cancer and is associated with the highest mortality. Where melanoma is mostly unresponsive to conventional therapies (e.g., chemotherapy), BRAF inhibitor treatment has shown improved therapeutic outcomes. Photodynamic therapy (PDT) relies on a light-activated compound to produce death-inducing amounts of reactive oxygen species (ROS). Their capacity to selectively accumulate in tumor cells has been confirmed in melanoma treatment with some encouraging results. However, this treatment approach has not reached clinical fruition for melanoma due to major limitations associated with the development of resistance and subsequent side effects. These adverse effects might be bypassed by immunotherapy in the form of antibody–drug conjugates (ADCs) relying on the ability of monoclonal antibodies (mAbs) to target specific tumor-associated antigens (TAAs) and to be used as carriers to specifically deliver cytotoxic warheads into corresponding tumor cells. Of late, the continued refinement of ADC therapeutic efficacy has given rise to photoimmunotherapy (PIT) (a light-sensitive compound conjugated to mAbs), which by virtue of requiring light activation only exerts its toxic effect on light-irradiated cells.
- Skin cancer
- Antibody-based immunotherapy
- Melanoma
1. Introduction
Melanoma represents the most aggressive, malignant phenotype resulting from a genetic and/or environmental-induced change to epidermal skin melanocytes and accounts for more than 75% of skin cancer-related deaths[1][2]. It mostly affects light-skinned individuals who are excessively exposed to solar ultraviolet radiation A and B (UVA and UVB), which are able to indirectly or directly cause DNA damage through oxidative (reactive oxygen species, ROS) or genotoxic stresses, respectively[3][4][5]. Alternatively, a genetic predisposition acquired through B-Raf proto-oncogene (BRAFV600E) mutation (present in more than 60% of melanoma patients) is characterized by the substitution of the amino acid aspartic acid by valine at position 600 and may lead to melanoma pathogenesis or melanomagenesis [6]. BRAFV600E induces constitutive kinase activity (e.g., mitogen-activated protein kinase pathway activation known as MAPK) which drives the uncontrolled growth of melanoma cells and pro-tumorigenic angiogenesis leading to disease metastases[6][7].
To date, the gold standard of therapy for malignant melanoma tumor is surgical resection[2]. Once melanomas reach the advanced metastatic stage, systemic therapies using the first US Food and Drug Administration (FDA)-approved chemotherapeutic drug dacarbazine (DTIC) and high-dose interleukin-2 (HD-IL-2, FDA-approved in 1998) have become the mainstay of treatments[8][9][10][11][12][13][14]. Unfortunately, the clinical success of these systemic therapies was hampered by severe dose-limiting toxicities, which did not improve overall patient survival [14][15]. In light of this, novel palliative treatment approaches were urgently needed to specifically treat patients with refractory and metastatic disease and to help circumvent these undesired toxicities to improve the overall therapeutic efficacy and patient survival.
2. Melanoma Immunotherapy
Conventional cancer therapies (e.g., surgery, radiation, and chemotherapy) have shown limited therapeutic benefits in patients with metastatic disease[16][17]. Despite significant advances in the development of systemic therapies, the therapeutic usage of toxic agents remains a double-edged sword, potentially causing side effects and restricting treatment to certain therapeutic dosages[18][19]. Consequently, novel therapeutic strategies were developed to specifically treat patients with recalcitrant metastases. Cancer immunotherapy in the form of adoptive cell therapy (ACT) has shown the capacity to represent such a therapy, with the ability to harness the patient’s own immunity against tumors[20][21][22]. In order to achieve maximum therapeutic efficacy, cancer immunotherapy relies on antigen recognition of tumor cells by cells of the innate immune system such as DCs (antigen-presenting cells (APCs)), which subsequently migrate to secondary lymphoid tissue to prime CTLs that are able to destroy tumors in an antigen-dependent manner[23][24]. These ACT attributes led to the FDA approval of sipuleucel-T (in 2010), which is a DC vaccine that is used for the treatment of asymptomatic or minimally symptomatic castration-resistant prostate cancer[25][26]. Sipuleucel-T is able to activate autologous anti-tumor immune reactions toward prostate tumors overexpressing prostatic acid phosphatase tumor antigens[25][27][28]. This DC-based vaccine (sipuleucel-T) achieved high objective response rates in melanoma patients (8–15%), which were characterized by an improved overall survival (20%) mediated through a robust CTL and natural killer cell (NK)-dependent immune response[29][30][31].
3. Antibody-Based Immunotherapy
In order to overcome ACT drawbacks, immunotherapeutic treatments were developed in a form of molecular-targeted therapies using mAbs. However, each mAb possesses an antigen-binding region known as a fragment variable region (Fab) and a constant region (fragment crystallizable: Fc domain) with an effector function (Figure 1). The Fv (fragment variable region) fragment of a mAb is made up of a variable light chain (VL) and a heavy chain (VH), containing three complementarity-determining regions (CDRs) and four framework regions (FRs)[32][33][34]. Traditionally, these mAbs exercise their cytotoxic effects through their Fc domain, within the constant region, which functions by interacting with immune effector cells and mediates tumor destruction via ADCC, CDC, or receptor blockade (Figure 1)[34][35][36]. Unfortunately, the success of this immunotherapeutic treatment (e.g., high-dose-IL-2) relies on high dosages and multiple treatment schedules, thus limiting clinical benefits[37][38][39][40]. Therefore, ipilimumab, a fully human mAb (immunoglobulin G1, or IgG1) that targets and blocks CTLA-4, was developed and clinically approved by the FDA as the first immune checkpoint inhibitor to treat metastatic melanoma patients[41][42][43]. CTLA-4 is a CD28 homolog, a T-lymphocyte co-stimulatory receptor that normally binds to cognate B7-ligand expressed on APCs such as DCs to activate T cell function[39][42][43]. Unfortunately, when CTLA-4 outcompetes CD28 for binding on a cognate B7 ligand, as a result of higher affinity and avidity, it activates T cells exhaustion, compromising antitumor immune responses[39][42][43]. Hence, by preventing CTLA-4 interaction with B7, ipilimumab acts to reinvigorate previously exhausted T-cells and boosts antitumor immunity through enhanced immune effector functions[39][42][43]. This therapeutic success (CTLA-4) spurred further development, leading to the FDA approval (in 2015) of new immune cell blockade (ICB) anti-PD-1 mAbs (pembrolizumab and nivolumab) binding respectively to their natural programmed death ligands 1/2 (PDL-1 and PDL-2) largely expressed on various immune cells (T cells, B cells, NK cells, macrophages, and DCs) and tumor cells [44][45]. For instance, nivolumab gained clinical approval following the Check-Mate006 clinical trials on patients with unresected and advanced melanoma[44]. During this study, nivolumab was shown to produce a progression-free survival (5.1 versus 2.2 months) and an objective response rate (40% versus 13.9%) superior to the DTIC-treated patients, respectively[44][46]. Similarly, pembrolizumab showed better therapeutic benefits, which were characterized by higher progression-free survival (e.g., 6 months in 47.3% of biweekly treated patients) and overall survival (e.g., 1 year survival in 74% biweekly treated patients) when compared to ipilimumab (e.g., 6 months in 26.5% and 58% of overall survival) [44][47]. In spite of their therapeutic successes (anti-CTLA-4 and anti-PD-1), only a subset of patients manifests a durable response[43][48]. Currently, a palliative approach is being tested in a phase III clinical trial (NCT02224781) combining ICB therapies (ipilimumab and nivolumab) with dabrafenib and trametinib (NCT02224781). This combinatorial approach was supporting Sanlorenzo et al. (2018) findings, showing how BRAFi/MEKi treatment could be synergized with anti-PD-1 therapy to kill BRAFV600E-positive melanoma tumor cells[49]. Another phase II clinical trial (NCT02908672) investigating the combination of atezolizumab (fully humanized anti-PDL-1) with cobimetinib (MEK inhibitor) and vemurafenib against vemurafenib and cobimetinib treatment is presently being tested on metastatic melanoma patients. Lately, a phase I clinical trial (NCT02967692) was designed to assess the safety and efficacy of the spartalizumab (anti-PD-1 mAb) combination with a BRAF inhibitor (dabrafenib) and an MEK inhibitor (trametinib) in unresectable or metastatic BRAFV600E mutants. The success of this antibody-based immunotherapeutic treatment has been limited by multiple factors: (1) non-specific biomarker selection leading to the identification of irrelevant TAAs, (2) inefficacy of mAbs to treat cancers, (3) reduced mAbs internalization into tumor tissues (due to their bulky size), (4) production of neutralizing antibodies (or anti-idiotypic antibody) against mAbs of human origin, (5) off-target effects and immunogenicity when used in humans with functional immune systems, limiting repeated treatment dosage schedules and (6) common sides effects such as fatigue, rash, skin disorders, endocrinopathies, diarrhea, pneumonitis, and colitis[50][51][52][53].
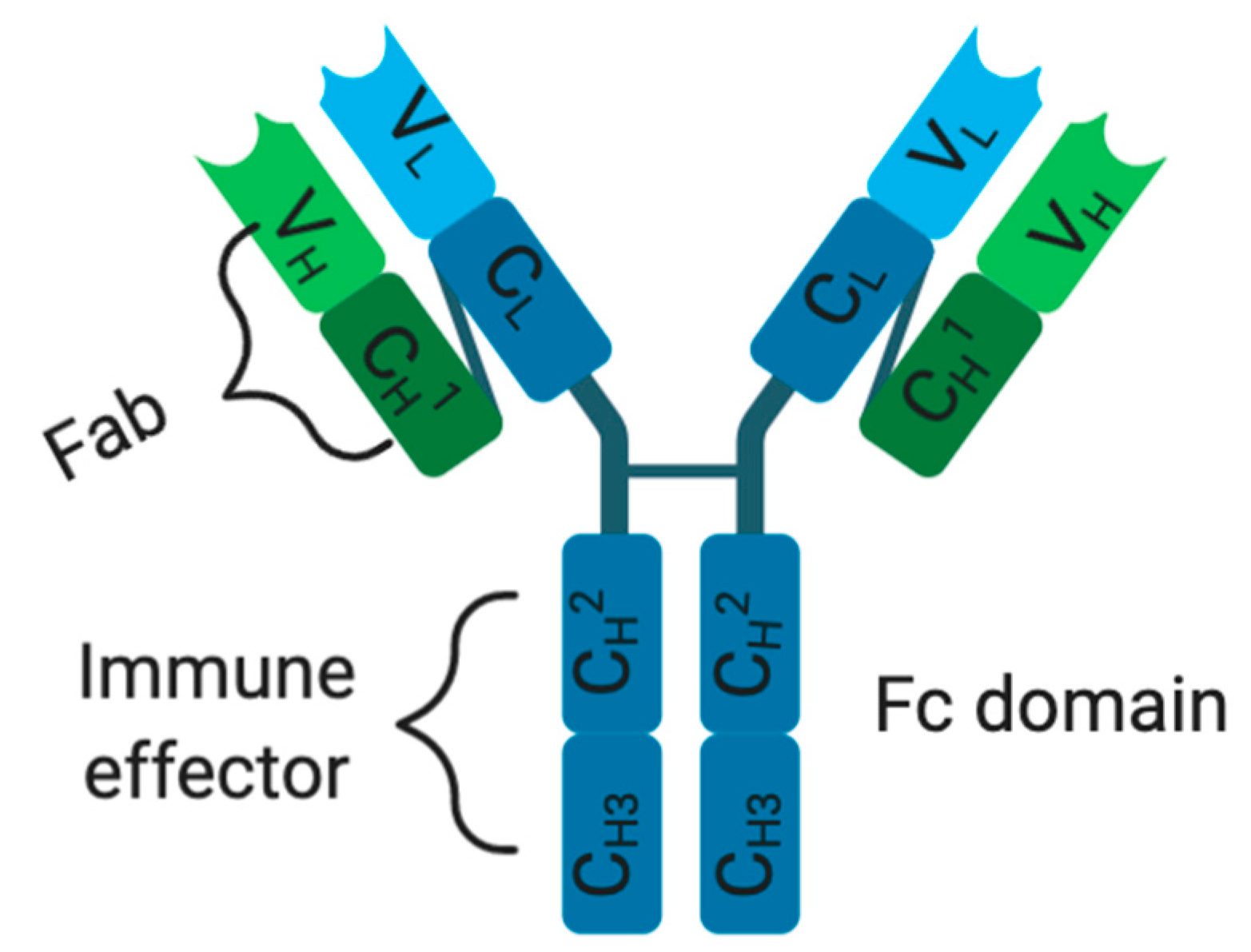 Figure 1. Monoclonal antibody structure. C: constant domain, V: variable domain, H and L: heavy and light chains.
Figure 1. Monoclonal antibody structure. C: constant domain, V: variable domain, H and L: heavy and light chains.
As such, despite early promise, the clinical application of therapeutic murine mAbs was severely hampered by their incapacity to efficiently activate human effector functions and their immunogenicity, which gave rise to human anti-mouse antibodies (HAMA)[50]. This immunogenic response toward the fully xenogeneic murine mAb led to poor therapeutic efficacy due to the neutralization and/or premature clearance of mAbs from the bloodstream, causing serious life-threatening side effects such as allergic and immune-mediated reactions (e.g., thyroiditis)[54][55]. This therapeutic inefficacy was demonstrated when cutaneous T-cell lymphoma (CTCL) and melanoma patients were treated with murine mAbs T101 and 9.2.27, respectively[56]. Half of the treated CTCL patients and three melanoma patients were shown to develop immunogenicity, which was characterized by an increased production of human antibodies against mouse immunoglobulin G (mIgG), especially with a repeated treatment cycle[56][57]. This immunogenic response is a significant problem, as it markedly compromised the widespread and repeated application of mAbs to treat various diseases. To mitigate these effects, recombinant protein technology was developed and led to the engineering of chimeric antibodies with ameliorated therapeutic outcomes[58]. These antibody formats consist of fusing an antigen-binding region (Fab: endowed with the antigen-binding capacity of mouse xenogeneic origin) to a human antibody Fc region possessing the effector functions that mediate ADCC[59]. These chimeric antibodies have very low levels of immunogenicity, enabling repeated dose treatment schedules with conserved efficacy of the parental mAb[32]. Rituximab (FDA-approved in 1997) is an example of a chimeric anti-CD20 mAb (consisting of a murine CD20 binding variable region of IgG1 mAb IDEC-2B8, which is genetically fused to a human IgG1 and kappa constant regions) used to treat multiple cancers[60]. Rituximab was shown to moderately improve therapeutic efficacy when treating melanoma patients[61]. In contrast, Velter et al. (2014) demonstrated that rituximab could worsen melanoma treatment or induce melanoma while treating B-cell lymphoma patients[62]. These results prompted the further optimization of mAbs, aiming at improving chimeric antibody properties by humanizing the fragment variable regions (Fab), which possess antigen-binding activity. Humanization of an antibody can be performed through various methods including the grafting of CDRs, veneering through surface manipulation of the framework region (FR) and transgenic mice using hybridoma technology. During the grafting method, xenogeneic VH and VL of the variable region sequence (CDRs) are joined to the human depleted CDR immunoglobulin scaffold[60][61]. Although this process drastically reduces the antigenicity of murine mAb in humans, it may alter the humanized antibody–antigen binding capacities, which in turn can influence its pharmacokinetic properties. The further improvement of mAbs can be achieved through the veneering method, which minimizes xenogeneic mAb antigenicity in human, by substituting xenogeneic FR-exposed residues with those mostly found in human antibodies. This is particularly relevant, as antigen-binding affinity relies heavily on the topography and chemical structure of the CDRs and some framework residues to maintain its binding affinity[63][64][65][66]. This was confirmed by Padlan (1991), who reported that human and rodent-derived immunoglobulin VH and VL possess unique features in exposed residues, which vary across the species[67][68]. Hence, an ideal antibody humanization should generate a product with (1) reduced immunogenicity and (2) conserved antigen-binding affinity on the non-human CDRs. To achieve these goals, humanization procedures should substitute exposed residues within the FR regions of the human scaffold with the murine exposed residues. This can be performed by selecting the human Fab region showing the greatest sequence homology to the specific murine Fab region consensus sequence[69]. Yet, few studies were able to simultaneously preserve the antigen-binding properties and reduce the murine-derived CDR-induced antigenicity by simply grafting the latter xenogeneic CDR to the human-depleted immunoglobulin[69]. These limitations paved the way to the development of transgenic mice, which enabled the production of a fully human antibody. These mice were engineered to possess functional human immunoglobulin transgenes, replacing their mouse orthologues, which are genetically inactivated[70][71]. In 1998, a humanized mAb gained FDA approval to treat human epidermal growth factor receptor 2 (HER2)-positive breast tumors[60][61][67][70]. Thereafter, ipilimumab (targeting CTLA-4, FDA-approved in 2011) and spartalizumab (humanized IgG4-PD-1) were developed to treat melanoma patients[72][73]. Indeed, while the clinical efficacy of DTIC in metastatic melanoma was low and did not offer any confirmed survival benefit, alternative treatment guidelines (based on the use of fully human mAbs) were being approved by the FDA[74][75]. For instance, patients with unresectable (advanced) stage III or IV melanoma received ipilimumab and nivolumab (targeting PD-1) as concurrent therapy, resulting in a 3-years overall survival rate of 63% in 94 patients[75]. As a result of the above-mentioned clinical successes, it became obvious that mAbs could be used as immunotherapeutic agents. Recently, antibody genetic engineering has permitted the production of genetically truncated versions, which are devoid of their effector Fc domain. These unnatural antibodies still retain their antigen-binding properties and can be generated through the randomization of CDRs of the Fab regions (Figure 3)[76][77]. Interestingly, these new antibody formats can be genetically or chemically fused to a fusion protein or cytotoxic agent to exert their potent effects as previously reported[61][72][78][79]. For example, a single-chain fragment (scFv) consisting of VH and VL chains of a mAb (about 30 kDa) linked by a short peptide sequence can be genetically fused using interdomain chains to form multivalent antibodies, such as diabody (60 kDa) or triabody (90 kDa), resulting in high-avidity properties[64]. However, the therapeutic activity of these novel antibodies will solely depend on the function of their conjugated warhead toxin or toxic agents. Based on these observations, it was quickly realized that naked mAbs against TAAs would not reach therapeutic fruition in existing pre-clinical animal models and that it would likely need to be coupled with toxic agents (e.g., small molecule toxins or PSs) to achieve improved anti-tumor responses.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines8090327
References
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.H.; Lok, H.C.; Sahni, S.; Lane, D.J.R.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Baldea, I.; Filip, A.G.; Napoca, C. Photodynamic therapy in melanoma an update. J. Physiol. Pharmacol. 2012, 63, 109–118. [Google Scholar] [PubMed]
- Mackie, R.M.; Hauschild, A.; Eggermont, A.M.M. Epidemiology of invasive cutaneous melanoma. Ann. Oncol. 2009, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chartrain, M.; Riond, J.; Stennevin, A.; Vandenberghe, I.; Gomes, B.; Lamant, L.; Meyer, N.; Gairin, J.E.; Guilbaud, N.; Annereau, J.P. Melanoma chemotherapy leads to the selection of ABCB5-expressing cells. PLoS ONE 2012, 7, e36762. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, D.L.; Saladi, R.N.; Fox, J.L. Ultraviolet radiation and skin cancer. Int. J. Dermatol. 2010, 49, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of us food and drug administration-approved targeted therapy in advanced melanoma. Onco Targets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [PubMed]
- Fedorenko, I.V.; Paraiso, K.H.T.; Smalley, K.S.M. Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem. Pharmacol. 2011, 82, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Biteghe, F.N.; Davids, L.M. A combination of photodynamic therapy and chemotherapy displays a differential cytotoxic effect on human metastatic melanoma cells. J. Photochem. Photobiol. B Biol. 2017, 166, 18–27. [Google Scholar] [CrossRef]
- Ugurel, S.; Paschen, A.; Becker, J.C. Dacarbazine in melanoma: From a chemotherapeutic drug to an immunomodulating agent. J. Investig. Dermatol. 2013, 133, 289–292. [Google Scholar] [CrossRef]
- Koprowska, K.; Hartman, M.L.; Sztiller-Sikorska, M.; Czyz, M.E. Parthenolide enhances dacarbazine activity against melanoma cells. Cancer Biol. Ther. 2013, 24, 835–845. [Google Scholar] [CrossRef]
- Marabondo, S.; Kaufman, H.L. High-dose interleukin-2 (IL-2) for the treatment of melanoma: Safety considerations and future directions. Expert Opin. Drug Saf. 2017, 12, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Ridolfi, L.; de Rosa, F.; Ridolfi, R.; Gentili, G.; Valmorri, L.; Scarpi, E.; Parisi, E.; Romeo, A.; Guidoboni, M. Radiotherapy as an immunological booster in patients with metastatic melanoma or renal cell carcinoma treated with high-dose Interleukin-2: Evaluation of biomarkers of immunologic and therapeutic response. J. Transl. Med. 2014, 12, 262. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Tykodi, S.S.; Thompson, J.A. Treatment of metastatic melanoma: An overview. Oncology (Williston Park) 2009, 23, 488–496. [Google Scholar] [PubMed]
- Palathinkal, D.M.; Sharma, T.R.; Koon, H.B.; Bordeaux, J.S. Current systemic therapies for melanoma. Dermatol. Surg. 2014, 40, 948–963. [Google Scholar] [CrossRef] [PubMed]
- Oh, A.; Tran, D.M.; McDowell, L.C.; Keyvani, D.; Barcelon, J.A.; Merino, O.; Wilson, L. Cost-effectiveness of nivolumab-ipilimumab combination therapy compared with monotherapy for first-line treatment of metastatic melanoma in the United States. J. Manag. Care Spec. Pharm. 2017, 23, 653–664.
- Nagaya, T.; Nakamura, Y.; Okuyama, S.; Ogata, F.; Maruoka, Y.; Choyke, P.L.; Kobayashi, H. Near-Infrared Photoimmunotherapy Targeting Prostate Cancer with Prostate-Specific Membrane Antigen (PSMA) Antibody. Cancer Res. 2017, 15, 1153–1162.
- Ogata, F.; Nagaya, T.; Nakamura, Y.; Sato, K. Near-infrared photoimmunotherapy: A comparison of light dosing schedules. Oncotarget 2017, 8, 35069.
- Kobayashi, H.; Griffiths, G.L.; Choyke, P.L. Near-Infrared Photoimmunotherapy: Photoactivatable Antibody–Drug Conjugates (ADCs). Chem. 2019.
- Zhang, H.; Ye, Z.L.; Yuan, Z.G.; Luo, Z.Q.; Jin, H.J.; Qian, Q.J. New strategies for the treatment of solid tumors with CAR-T cells. J. Biol. Sci. 2016, 12, 718–729.
- Ndhundhuma, I.M.; Abrahamse, H. Susceptibility of In Vitro Melanoma Skin Cancer to Photoactivated Hypericin versus Aluminium(III) Phthalocyanine Chloride Tetrasulphonate. Biomed. Res. Int. 2017, 2017, 5407012. [Google Scholar] [CrossRef]
- Davids, L.M.; Kleemann, B.; Kacerovská, D.; Pizinger, K.; Kidson, S.H. Hypericin phototoxicity induces different modes of cell death in melanoma and human skin cells. J. Photochem. Photobiol. B Biol. 2008, 91, 67–76. [Google Scholar] [CrossRef]
- Maduray, K.; Odhav, B.; Nyokong, T. In vitro photodynamic effect of aluminum tetrasulfophthalocyanines on melanoma skin cancer and healthy normal skin cells. Photodiagnosis Photodyn. Ther. 2012, 9, 32–39.
- Kawczyk-Krupka, A.; Bugaj, A.M.; Latos, W.; Zaremba, K.; Sieroń, A. Photodynamic therapy in treatment of cutaneous and choroidal melanoma. Photodiagnosis Photodyn. Ther. 2013, 10, 503–509.
- Ryabaya, O.O.; Inshakov, A.N.; Egorova, A.V.; Emelyanova, M.A.; Nasedkina, T.V.; Zasedatelev, A.S.; Khochenkov, D.A.; Stepanova, E.V. Autophagy inhibitors chloroquine and LY294002 enhance temozolomide cytotoxicity on cutaneous melanoma cell lines in vitro. Anticancer Drugs 2017, 3, 307–315.
- Yang, J.; Shangguan, J.; Eresen, A.; Li, Y.; Wang, J.; Zhang, Z. Dendritic cells in pancreatic cancer immunotherapy: Vaccines and combination immunotherapies. Pathol. Res. Pract. 2019, 215, 152691. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Geskin, L.J.; Damiano, J.J.; Patrone, C.C.; Butterfield, L.H.; Kirkwood, J.M.; Falo, L.D. Three antigen-loading methods in dendritic cell vaccines for metastatic melanoma. Melanoma Res. 2018, 28, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Eagles, M.E.; Nassiri, F.; Badhiwala, J.H.; Suppiah, S.; Almenawer, S.A.; Zadeh, G.; Aldape, K.D. Dendritic cell vaccines for high-grade gliomas. Ther. Clin. Risk Manag. 2018, 14, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Bae, Y.-S. Dendritic cell-based therapeutic cancer vaccines: Past, present and future. Clin. Exp. Vaccine Res. 2014, 3, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Smits, E.L.; Lion, E.; Van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267.
- Ross, J.S.; Gray, K.; Gray, G.S.; Worland, P.J.; Rolfe, M. Anticancer antibodies. J. Clin. Pathol. 2003, 119, 472–485.
- Safdari, Y.; Farajnia, S.; Asgharzadeh, M.; Khalili, M. Antibody humanization methods—A review and update. Genet. Eng. Rev. 2013, 29, 175–186.
- Peters, S.; Kerr, K.M.; Stahel, R. PD-1 blockade in advanced NSCLC: A focus on pembrolizumab. Cancer Treat. Rev. 2018, 62, 39–49.
- Morisada, M.; Clavijo, P.E.; Moore, E.; Sun, L.; Chamberlin, M.; Van Waes, C.; Hodge, J.W.; Mitchell, J.B.; Friedman, J.; Allen, C.T. PD-1 blockade reverses adaptive immune resistance induced by high-dose hypofractionated but not low-dose daily fractionated radiation. Oncoimmunology 2018, 7.
- Hudson, P.J. Recombinant antibody constructs in cancer therapy. Opin. Immunol. 1999, 11, 548–557.
- Marabondo, S.; Kaufman, H.L. High-dose interleukin-2 (IL-2) for the treatment of melanoma: Safety considerations and future directions. Expert Opin. Drug Saf. 2017, 12, 1347–1357.
- Michaelis, M.; Rothweiler, F.; Nerreter, T.; Van Rikxoort, M.; Sharifi, M.; Wiese, M.; Ghafourian, T.; Cinatl, J. Differential effects of the oncogenic BRAF inhibitor PLX4032 (vemurafenib) and its progenitor PLX4720 on ABCB1 function. Pharm. Pharm. Sci. 2014, 17, 154–168.
- Boudewijns, S.; Koornstra, R.H.T.; Westdorp, H.; Schreibelt, G.; Eertwegh, A.J.M.V.D.; Foppen, M.H.G.; Haanen, J.B.; De Vries, I.J.M.; Figdor, C.G.; Bol, K.F.; et al. Ipilimumab administered to metastatic melanoma patients who progressed after dendritic cell vaccination. OncoImmunology 2016, 5, e1201625. [Google Scholar] [CrossRef]
- Huang, M.-N.; Nicholson, L.T.; Batich, K.A.; Swartz, A.M.; Kopin, D.; Wellford, S.; Prabhakar, V.K.; Woroniecka, K.; Nair, S.K.; E Fecci, P.; et al. Antigen-loaded monocyte administration induces potent therapeutic antitumor T cell responses. J. Clin. Investig. 2020, 130, 774–788.
- Khair, D.O.; Bax, H.J.; Mele, S.; Crescioli, S.; Pellizzari, G.; Khiabany, A.; Nakamura, M.; Harris, R.J.; French, E.; Hoffmann, R.M.; et al. Combining immune checkpoint inhibitors: Established and emerging targets and strategies to improve outcomes in melanoma. Immunol. 2019, 10, 453.
- Letendre, P.; Monga, V.V.; Milhem, M.M.; Zakharia, Y. Ipilimumab: From preclinical development to future clinical perspectives in melanoma. Future Oncol. 2017, 13, 625–636.
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Mol. Med. 2018, 50, 1–11.
- Franklin, C.; Livingstone, E.; Roesch, A.; Schilling, B.; Schadendorf, D. Immunotherapy in melanoma: Recent advances and future directions. J. Surg. Oncol. (EJSO) 2017, 43, 604–611.
- Xu-Monette, Z.Y.; Zhang, M.; Li, J.; Young, K.H. PD-1/PD-L1 Blockade: Have We Found the Key to Unleash the Antitumor Immune Response? Immunol. 2017, 8.
- Robert, C.; Long, G.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. Engl. J. Med. 2015, 372, 320–330.
- Robert, C.; Schachter, J.; Long, G.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. Engl. J. Med. 2015, 372, 2521–2532.
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330.
- Sanlorenzo, M.; Vujic, I.; Floris, A.; Novelli, M.; Gammaitoni, L.; Giraudo, L.; Macagno, M.; Leuci, V.; Rotolo, R.; Donini, C.; et al. BRAF and MEK Inhibitors Increase PD-1-Positive Melanoma Cells Leading to a Potential Lymphocyte-Independent Synergism with Anti–PD-1 Antibody. Cancer Res. 2018, 24, 3377–3385.
- Waldmann, H. Human monoclonal antibodies: The benefits of humanization. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2019; Volume 1904, pp. 1–10. [Google Scholar]
- Brennan, F.R.; Morton, L.D.; Spindeldreher, S.; Kiessling, A.; Allenspach, R.; Hey, A.; Muller, P.Y.; Frings, W.; Sims, J. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. MAbs 2010, 2, 233–255. [Google Scholar] [CrossRef]
- Weiner, L.M.; Dhodapkar, M.V.; Ferrone, S. Monoclonal Antibodies for Cancer Immunotherapy. Lancet 2009, 373, 1033–1040. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: Mechanism, combinations, and clinical outcome. Front. Pharmacol. 2017, 8, 561.
- Green, L.L. Transgenic Mouse Strains as Platforms for the Successful Discovery and Development of Human Therapeutic Monoclonal Antibodies. Curr. Drug Discov. Technol. 2014, 11, 74–84. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Goyal, A.; Kaur, P.; Singh, R.; Kalra, S. Anticancer Drug-induced Thyroid Dysfunction. Eur. Endocrinol. 2020, 16, 32–39.
- Igg, M.; Shawler, D.L.; Bartholomew, R.M.; Linda, M.; Dillman, R. Human immune response to multiple injections of murine monoclonal IgG. Immunol. 1985, 135, 1530–1535.
- Schroff, R.W.; Foon, K.A.; Beatty, S.M.; Oldham, R.K.; Morgan, A.C. Human Anti-Murine Immunoglobulin Responses in Patients Receiving Monoclonal Antibody Therapy. Cancer Res. 1985, 45, 879–885.
- Maloney, D.G. Preclinical and phase I and II trials of rituximab. Oncol. 1999, 26, 74–78.
- Press, O.W. Radiolabeled Antibody Therapy of B-Cell Lymphomas. Oncol. 1999, 26, 58–65.
- Winkler, J.K.; Schiller, M.; Bender, C.; Enk, A.H.; Hassel, J.C. Rituximab as a therapeutic option for patients with advanced melanoma. Cancer Immunol. Immunother. 2018, 67, 917–924.
- Velter, C.; Pagès, C.; Schneider, P.; Osio, A.; Brice, P.; Lebbé, C. Four cases of rituximab-associated melanoma. Melanoma Res. 2014, 24, 401–403.
- Padlan, E.A. Anatomy of the antibody molecule. Mol. Immunol. 1994, 31, 169–217. [Google Scholar] [CrossRef]
- Welschof, M.; Krauss, J. Recombinant Antibodies for Cancer Therapy; Humana: Totowa, NJ, USA, 2003; ISBN 0896039188. [Google Scholar]
- Queen, C.; Schneider, W.P.; Selick, H.E.; Payne, P.W.; Landolfi, N.F.; Duncan, J.F.; Avdalovic, N.M.; Levitt, M.; Junghans, R.P.; Waldmann, T.A. A humanized antibody that binds to the interleukin 2 receptor. Proc. Natl. Acad. Sci. USA 1989, 86, 10029–10033. [Google Scholar] [CrossRef]
- Co, M.S.; Queen, C. Humanized antibodies for therapy. Nature 1991, 351, 501–502. [Google Scholar] [CrossRef]
- Padlan, E.A. A possible procedure for reducing the immunogenicity of antibody variable domains while preserving their ligand-binding properties. Mol. Immunol. 1991, 28, 489–498. [Google Scholar] [CrossRef]
- Pedersen, J.T.; Henry, A.H.; Searle, S.J.; Guild, B.C.; Roguska, M.; Rees, A.R. Comparison of surface accessible residues in human and murine immunoglobulin Fv domains. Implication for humanization of murine antibodies. J. Mol. Biol. 1994, 235, 959–973. [Google Scholar] [CrossRef]
- Roguska, M.A.; Pedersen, J.T.; Henry, A.H.; Searle, S.M.; Roja, M.; Avery, B.; Hoffee, M.; Cook, S.; Lambert, J.M.; Blattler, W.A.; et al. A comparison of two murine monoclonal antibodies humanized by CDR-grafting and variable domain resurfacing. Protein Eng. 1996, 9, 895–904. [Google Scholar] [CrossRef]
- DM, F.; Sl, O.D.; Bengoechea, T.; Dv, H.; Harding, F.; Sl, B.; Rm, K.; Km, H.; Sr, S.; Lonberg, N. High-avidity human IgG kappa monoclonal antibodies from a novel strain of minilocus transgenic mice. Nat. Biotechnol. 1996, 14, 845–851. [Google Scholar]
- Mendez, M.J.; Green, L.L.; Corvalan, J.R.; Jia, X.-C.; Maynard-Currie, C.E.; Yang, X.-D.; Gallo, M.L.; Louie, D.M.; Lee, D.V.; Erickson, K.L.; et al. Functional transplant of megabase human immunoglobulin loci recapitulates human antibody response in mice. Nat. Genet. 1997, 15, 146–156.
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. MAbs 2019, 11, 219–238.
- Malas, S.; Harrasser, M.; Lacy, K.E.; Karagiannis, S.N. Antibody therapies for melanoma: New and emerging, opportunities to activate immunity (Review). Rep. 2014, 32, 875–886.
- Albertini, M.R. The age of enlightenment in melanoma immunotherapy. Immunother. Cancer 2018, 6, 80.
- Callahan, M.K.; Kluger, H.; Postow, M.A.; Segal, N.H.; Lesokhin, A.; Atkins, M.B.; Kirkwood, J.M.; Krishnan, S.; Bhore, R.; Horak, C.; et al. Nivolumab plus ipilimumab in patients with advanced melanoma: Updated survival, response, and safety data in a phase i dose-escalation study. Clin. Oncol. 2018, 36, 391–398.
- Choromańska, A.; Saczko, J.; Kulbacka, J.; Skolucka, N.; Majkowski, M. The potential role of photodynamic therapy in the treatment of malignant melanoma—An in vitro study. Clin. Exp. Med. 2012, 21, 179–185.
- Beatty, G.L.; Moon, E.K. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. Oncoimmunology 2014, 3, e970027.
- Amoury, M.; Mladenov, R.; Nachreiner, T.; Pham, A.T.; Hristodorov, D.; Di Fiore, S.; Helfrich, W.; Pardo, A.; Fey, G.; Schwenkert, M.; et al. A novel approach for targeted elimination of CSPG4-positive triple-negative breast cancer cells using a MAP tau-based fusion protein. J. Cancer 2016, 139, 916–927.
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.B.; Henner, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Natl. Acad. Sci. USA 1992, 89, 4285–4289.
- Sanz, L.; Cuesta, Á.M.; Compte, M.; Álvarez-Vallina, L. Antibody engineering: Facing new challenges in cancer therapy. Acta Pharmacol. Sin. 2005, 26, 641–648.