2. Nutritional and Lifestyle Aspects That Increase Susceptibility to Gallbladder Carcinogenesis
As previously mentioned, gallstone disease (GSD) is one of the most important predisposing factors to developing GBC. Thus, metabolic syndrome and some factors that increase the probability of forming gallstones, including sedentary lifestyle, the consumption of sugar-sweetened and artificially sweetened beverages, obesity, high-fat diet, hypercholesterolemia, and the consumption of red meat, can consequently promote carcinogenesis by inducing a chronic pro-inflammatory state in the gallbladder tissue [
2,
6,
27,
93]. Conversely, a physically active lifestyle and the consumption of some vegetables, such as radish and sweet potato, have been shown to reduce the risk of developing gallstones and GBC [
27,
94]. In this regard, an increased BMI constitutes an important risk factor for GBC and other types of cancer [
21,
95]. For instance, a study by Barahona et al. showed that BMI has a causal effect on gallstone disease, which subsequently increases the GBC risk. The meta-analysis performed by Tan et al. showed that the relative risk of GBC was 1.14 (95% CI, 1.04–1.25) for overweight people (BMI 25–30 kg/m
2) and 1.56 (95% CI, 1.41–1.73) for obese individuals (BMI > 30 kg/m
2), evidencing a higher GBC risk in women than men [
22]. This difference between genders could be explained by the fact that estrogens promote a greater cholesterol storage in the bile, which is consistent with the higher obesity and BMI rates observed in women, and the increased risk of developing gallstones [
96]. Interestingly, higher C-reactive protein (CRP) concentrations increased GBC risk in the European population, suggesting that the inflammatory status is crucial in developing GBC [
36].
The matter of a sedentary lifestyle can be explained according to metabolic disorders inferred from the study by Skoumas et al., in which physically active women had significantly lower levels of total serum cholesterol, LDL-c, oxidized LDL cholesterol, and triglycerides compared to sedentary women [
94]. Other studies have also observed that a more sedentary lifestyle is directly correlated with the risk of developing metabolic syndrome and elevated plasma levels of triglycerides and cholesterol [
97,
98]. On the other hand, the consumption of sugar-sweetened and artificially sweetened beverages can significantly increase (double) the risk of GBC when individuals consume two or more servings per day (200 mL/serving) of sweetened beverages compared to those without consumption (hazard ratio = 2.24; 95% CI = 1.02 to 4.89) [
18]. A probable explanation for this is that the increased sugar intake has been associated with increased body weight and diabetes mellitus, predisposing to gallstone disease-dependent GBC [
19]. In fact, diabetic patients have shown a higher risk of GBC compared to non-diabetic individuals [
13]. This phenomenon is probably due to the fact that hyperinsulinemia caused by sugar ingestion induces the expression of the insulin-like growth factor receptor (IGFR-1), which is able to induce malignant properties in several types of cancer by increasing cell proliferation, tumorigenic capacity, and apoptosis resistance through the activation of PI3K and MAPK pathways [
99].
In general, these data suggest that a sedentary lifestyle and sugar consumption—powered by high-fat food—can promote gallstone formation and subsequently GBC.
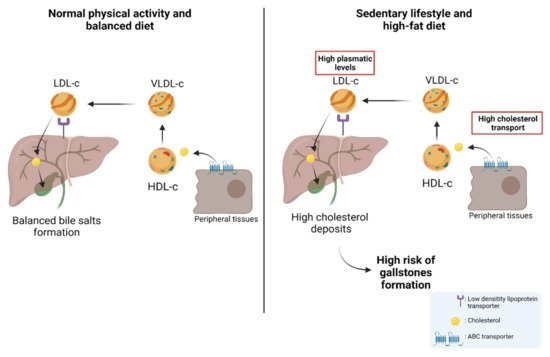
About 80% of gallstones are comprised of cholesterol; hence, patients with permanently high levels of plasma cholesterol are candidates for gallstones [
100,
101,
102,
103]. Physiologically, cholesterol is transported through different lipoproteins such as high-density lipoproteins (HDL-c) and low-density lipoproteins (LDL-c). HDL-c takes the cholesterol from peripheral tissues through ABC transporters (e.g., ABCA1) and esterifies it through lecithin cholesterol acyltransferase (LCAT) [
104]. Subsequently, HDL-c can be recognized by the scavenger receptor class B type I (SR-BI), which facilitates the uptake of high-density lipoprotein cholesterol esters in the liver. In addition, cholesterol can be transported to very-low-density lipoproteins (VLDL-c) via the cholesteryl ester transfer protein (CETP) [
105,
106]. Then, VLDL-c is transformed into LDL-c and is recognized by the low-density lipoprotein receptor (LDLr) and LDL-related protein (LRP) to induce the internalization of cholesterol into the hepatocytes in the liver [
105,
107]. Finally, the cholesterol within the hepatocytes can be used in bile formation to be transported to the bile canaliculus and stored in the gallbladder [
105,
108]. Interestingly, a study by Selcuk Atamanalp et al. showed that high cholesterol and LDL-c levels in plasma were closely correlated with a higher presence of cholesterol gallstones. Conversely, low serum HDL levels did not affect the occurrence of cholesterol gallstones [
109]. In addition, Wang et al. showed that patients with increased plasma levels of cholesterol, triglycerides, HDL-c, LDL-c, and apolipoprotein B (APOB), also showed a significantly higher recurrence of gallstones compared to control patients [
110], which was similar to those results found by Hayat et al., who showed that patients with gallstones had significantly higher plasma levels of triglycerides and HDL-c than the control patients [
20]. All these data reaffirm the idea that high plasma levels of cholesterol and other lipids induce gallstone formation and, therefore, are heavily involved in the subsequent development of GBC. The cholesterol metabolism and its implication in the risk of developing GSD are represented in
Figure 1.
Figure 1. The cholesterol metabolism and the risk of developing gallstones. Left: Under normal conditions, cholesterol stored in peripheral tissues is transported to HDL-c via ABC transporters. The circulating HDL-c in the blood transports cholesterol to VLDL-c which is then transformed into LDL-c. Then, LDL-c transports cholesterol to the hepatocytes through a low-density lipoprotein receptor (LDLr). Cholesterol can be used in bile formation and stored in the gallbladder. Right: As a result of a high-fat diet and sedentary lifestyle, the concentration of plasma cholesterol increases, provoking a greater presence of cholesterol in peripheral tissues. This causes increased transportation of cholesterol from peripheral tissues to the liver via the different lipoproteins (HDL-c, VLDL-c, and LDL-c). Finally, this induces an increase in the storage of cholesterol in the liver and subsequently a greater release of cholesterol from the liver to the gallbladder, which leads to a high risk of gallstone formation.
There are different pathophysiological processes that promote gallstone formation, including cholesterol crystallization/nucleation, alteration in mucin secretion, changes in biliary motility, alteration in intestinal cholesterol transport, and intestinal motility [
93]. Normally, cholesterol is found in unilamellar vesicles that bind bile salts to allow cholesterol solubilization. However, when there is an increase in cholesterol deposits, the solubilizing capacity of the bile is saturated promoting the solidification and subsequent nucleation of cholesterol/bile acids, allowing the formation of gallstones [
111]. The mechanism by which cholelithiasis predisposes to GBC has not yet been established; however, it has been reported that the DNA mutation rate is higher in the inflammation microenvironment induced by gallstones than in normal tissues, increasing the risk of developing GBC [
112]. This inflammatory state is generated in different ways, including the direct damage by gallstones due to continuous friction with the mucosa, which leads to a continuous damage/repair loop in the gallbladder epithelium and, on the other hand, the increase in gallstone size which can obstruct the bile duct, which also increases the susceptibility to infections that give rise to the inflammation process [
93,
113].
Different animal models have been used to study gallstone formation and its effect on the development of preneoplastic lesions [
114,
115,
116,
117]. Our research group established an animal model of gallbladder preneoplasia through a lithogenic diet. The results showed that after administration of a high-cholesterol diet (lithogenic diet) for 9 months, early gallstone formation was induced within the gallbladder. Furthermore, animals fed a lithogenic diet evidenced fatty liver and higher plasma cholesterol levels than animals fed a normal diet in a similar manner to that observed in humans. More interestingly, those animals treated with a lithogenic diet had metaplastic and dysplastic architecture in gallbladder tissues, with no invasive features, but with a well-defined inflammatory component showing a predominance of lymphocytes and polymorphonuclear cells [
116]. A similar study showed that mice treated with a lithogenic diet had epithelial hyperplasia along with the occurrence of acute and chronic inflammation characterized by the presence of eosinophils, neutrophils, and lymphocytes within the lamina propria [
117]. Recently, Kato et al. described an animal model generated by the orthotopic implantation of gallbladder organoids containing mutant loss of
KRAS and
TP53 genes developed in vitro using lentiviral Cre transduction and CRISPR/Cas9 gene editing, respectively. The data showed that the tumor transcriptomic profiles are similar to that found in human tissues, as well as the immune cell infiltration observed during tumor formation, suggesting that this model could be an interesting approach to study carcinogenesis in GBC [
118].
Different inflammatory components have been described as participants in the pathophysiology of inflammation including some inflammatory cytokines such as IL-6, IL-10, IL-12, and visfatin. Interestingly, visfatin is considered a key molecule in the activation of human leukocytes and the production of pro-inflammatory cytokines; therefore, it seems to be associated with the risk of developing gallstones [
119,
120]. In this regard, patients with acute cholecystitis often present a higher expression of visfatin in peripheral blood mononuclear cells (PBMCs), serum, and in grossly inflamed gallbladder tissues. Moreover, the gene overexpression of visfatin observed on in vitro models of acute cholecystitis has been frequently accompanied by an increased expression of other pro-inflammatory mediators including IL-10, TNF-α, IL-6, ICAM-1, and VCAM-1 [
121]. Similarly, Nien Wang et al. showed that serum visfatin levels were markedly higher in subjects that presented pigment and cholesterol gallstones than in healthy controls. Additionally, serum levels of cholesterol, triglycerides, AST, ALT, leukocyte count, and fasting glucose were significantly higher in those individuals with gallstones. Interestingly, high AST levels and the increased white blood cell count were considered significant predictors of gallbladder lithiasis, while the elevated values of visfatin in serum were also suggested as a significant risk factor for gallstone formation [
120]. These results suggested that visfatin could be a predictive marker of inflammation and predisposition to gallbladder lithiasis.
As previously mentioned, evidence accumulated over many years indicates that gallstones can induce an inflammatory microenvironment that increases the risk of developing GBC. However, this risk can be fostered by a greater genetic predisposition [
122], for example, the genetic variability present in genes that encode the different ATP-binding cassette (ABC) transporters in the hepatocanalicular membrane, which are involved in the different processes of the exportation of bile salts in biliary tracts, including the transportation of ABCB11, the transport of phosphatidylcholine (ABCB4), and secretion of cholesterol and phytosterols into bile (heterodimer ABCG5/8) [
123]. In this regard, a genetic variant in the
ABCG8 gene (variant rs11887534 or D19H) has been associated with a higher gallstone development through cholesterol hypersecretion and cholesterol supersaturation in the bile [
122,
124]. In addition, other variants (rs1558375, rs17209837, and rs4148808) have been determined in the 7q21.12 region harboring both the
ABCB1 and
ABCB4 genes, which showed a higher risk of developing GBC [
28]. A study by Bustos et al. showed that in the Chilean Mapuche ancestry population, variants in
ABCG8 (rs11887534) and
TRAF3 (rs12882491) were associated with GSD. In addition, it was shown that TRAF3 levels were lower in individuals affected by GSD, suggesting that these variants could be used as risk markers for GBC [
125,
126]. Other mutations occur in the
ABCB4 gene and are classified as nonsense mutations (class I), missense mutations affecting maturation (class II), activity (class III), or protein stability (class IV), and mutations with no identifiable effect (class V) [
127]. It has been shown that mutations in this gene increase the risk of developing gallstones in subjects under 40 years, mainly by inducing an
ABCB4 deficiency that results in low biliary phosphatidylcholine concentrations, which is consistent with the spontaneous occurrence of cholecystolithiasis [
128,
129]. In fact, the homozygous
ABCB4 mutations lead to the complete absence of the phospholipid transporter and no secretion of phospholipids into bile, which finally causes a decrease in the solubility of bile, and consequently, a greater predisposition to bile crystallization and gallstone formation [
130]. Other mutations and alterations have been described in different genes and proteins potentially involved in a higher risk of developing gallstones, such as ABCB11 [
130], cholesterol 7a-hydroxylase (CYPA1) [
131,
132],
APOB gene [
133], and cholecystokinin A receptor (CCKAR) [
134,
135]. In addition, certain alterations in genes related to the immune system, inflammation, and oxidative stress have also been implicated in a greater risk of developing GBC, including mutations in
TLR2, TLR4 [
136],
IL1RN, IL1B [
137],
IL10, IL8, IL8RB, RNASEL, VEGF [
138], and
CCR5 [
139], as well as rs7504990 variant in the
DCC [
140]. These data show that the predisposition to developing cholecystolithiasis and GBC also have an important genetic background that needs to be considered in GBC carcinogenesis.
Finally, another risk factor associated with GBC is the appearance of bluish and brittle calcifications in the inner gallbladder wall named "porcelain gallbladder" [
141]. Porcelain gallbladder has an incidence of less than 1% in patients with gallbladder disease, being more prevalent in women [
141]. This rare condition is considered a risk factor for GBC because approximately 60% to 90% of these cases show gallstones [
141,
142]. Despite the pathophysiology of "porcelain gallbladder" not being clear, this condition could be a consequence of a previous chronic inflammatory process or could be the result of an obstruction produced by gallstones that induce the accumulation and precipitation of calcium in the mucosal layer of the gallbladder wall [
143]. Whatever the origin of this disease, it is also unknown whether calcium levels play a role in the “porcelain gallbladder” pathogenesis. Recently, Berger et al. found significantly higher calcium and parathormone (PTH) levels in the plasma of individuals with porcelain gallbladder compared to controls [
144], which suggests that individuals with diseases that induce persistent hypercalcemia (e.g., primary hyperparathyroidism) also have a higher risk of developing porcelain gallbladder. In this regard, we venture to propose that persistent hypercalcemia could be an initiating factor of porcelain gallbladder, which would eventually trigger the formation of a chronic inflammatory state in the inner layer of the gallbladder, increasing in this manner the risk of developing GBC. However, more studies are still needed to demonstrate this hypothesis.
Figure 3 shows the relationship between the risk factors described above and their involvement in GBC inflammation and carcinogenesis.
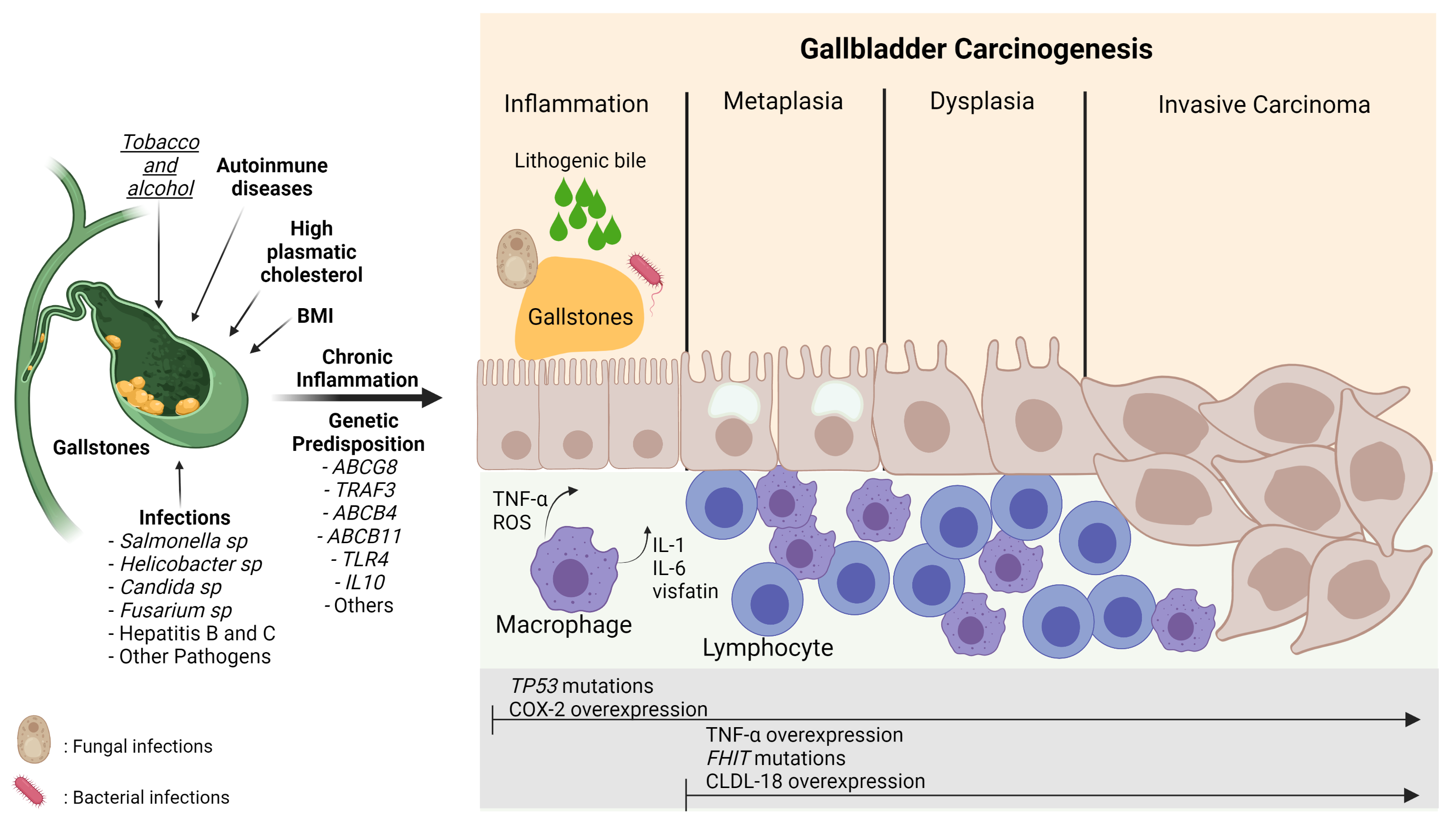
Figure 3. Carcinogenesis process in gallbladder cancer. The damage caused by the presence of risk factors such as gallstones, infections, lithogenic bile, alcohol, smoking, and genetic predisposition can induce continuous damage in the mucosa of the gallbladder, which is characterized by a chronic inflammatory state mainly highlighted by the activation of macrophages and lymphocytes that leads to the release of pro-inflammatory cytokines (TNF-α, IL-6, IL-1) and ROS stimulating the carcinogenic metaplasia/hyperplasia–dysplasia–carcinoma transition. This process can be marked by different gene alterations and protein expressions such asTP53andFHITmutations and COX-2, TNF-α, and CLDN-18 overexpression, respectively. BMI: Body Mass Index; TNF-α: Tumor necrosis factor-alpha; ROS: Reactive oxygen species; IL-1: Interleukin-1; IL-6; Interleukin-6; CLDN-18: Claudin18; COX-2: Cyclooxygenase 2; TP53: Tumor protein 53; FHIT: Fragile Histidine Triad Diadenosine Triphosphatase. The risk factors in bold mean strong evidence. The risk factor in italics and underlined means weak evidence