Non-alcoholic fatty liver disease (NAFLD) includes a range of chronic conditions characterized by excessive hepatic lipid accumulation, defined by the presence of steatosis in >5% of hepatocytes, in the absence of significant alcohol consumption or other causes of liver injury.
- non-alcoholic fatty liver disease
- non-alcoholic steatohepatitis
- chronic liver disease
- metabolic disorders
- hepatoprotection
- pharmacotherapy
1. Introduction
2. THRβ Agonists
3. Lipogenesis Inhibitors
3.1. ACC Inhibitors
3.2. FASN Inhibitors
3.3. SCD1 Inhibitors
3.4. DGAT Inhibitors
3.5. ω-3 PUFAs
4. Bile Acid Metabolism Modulators
4.1. FXR Agonists
4.2. FGF Analogues
5. Fibrogenesis Inhibitors
5.1. Galectin Antagonists
5.2. TLR4 Antagonists
5.3. LOXL2 Inhibitors
5.4. ATX Inhibitors
6. Glucose Metabolism Modulators
6.1. PPAR Agonists
6.2. MPC Inhibitors
In addition to their primary mechanism of action, pioglitazone and other thiazolidinediones are known to interact with the mitochondrial pyruvate carrier (MPC) to suppress pyruvate transport into the mitochondrial matrix. Since this direct, non-genomic effect is considered essential for the inhibition of hepatic gluconeogenesis by PPARγ ligands, novel MPC-inhibiting thiazolidinedione derivatives with minimal affinity towards PPARγ have been synthesized.
Azemiglitazone (MSDK-0602K) reduced liver enzyme levels in NASH/fibrosis patients regardless of T2DM presence [105], and was characterized by a markedly improved safety profile [106]. However, the drug did not demonstrate significant effects on any of the histological endpoints [105]. A phase 3 trial to evaluate azemiglitazone in diabetic or prediabetic patients with NAFLD/NASH has been initiated (NCT03970031). An alternative approach to minimizing the side effects of PPARγ agonists is represented by PXL065, the deuterium-stabilized R-isomer of pioglitazone [107], which is currently being assessed in a phase 2 RCT (NCT04321343).
6.3. Incretin Mimetics
Incretins are a small family of intestinal L-cell-derived peptide hormones that includes glucagon-like peptide 1 (GLP1), glucose-dependent insulinotropic polypeptide (GIP), and oxyntomodulin. The incretin axis mediates the physiological response to hyperglycaemia and couples glucose intake with pancreatic secretion [108]. GLP1 receptors (GLP1R) are expressed in the β-cells, hepatocytes, white adipose tissue, brain, and skeletal muscle [109]. GLP1R activation induces insulin secretion, decreases insulin resistance, inhibits glucagon release and lipogenesis, and suppresses appetite and gastrointestinal motility. Additionally, hepatic GLP1R stimulate FA β-oxidation, inhibit profibrogenic signaling pathways, and exert a mild anti-inflammatory effect by indirectly reducing CRP, proinflammatory cytokine, and chemokine production [110].
At the moment, semaglutide is the only GLP1R agonist being developed for the treatment of NASH in nondiabetic subjects. In the recently completed 72-week phase 2 trial, semaglutide treatment (0.4 mg/d) led to NASH resolution with no worsening of fibrosis in 59% vs. 17% in the placebo group, which is considered the highest response rate that a drug has ever achieved in a NASH trial up to now [111][112]. Currently, semaglutide is being evaluated in several phase 2 trials as monotherapy (NCT03884075, NCT04216589) as well as in combinations with empagliflozin (NCT04639414), and cilofexor/firsocostat (NCT04971785). A 5-year-long phase 3 study designed to include 1200 patients with precirrhotic NASH has been initiated (NCT04822181).
Exenatide, lixisenatide, liraglutide, and dulaglutide have all demonstrated significant antisteatotic activity, and (with the exception of lixisenatide) improved intrahepatic cholestasis in T2DM/NAFLD patients. Amelioration of cytolysis markers was reported for exenatide [113][114] and dulaglutide [115][116], and lixisenatide was effective against liver fibrosis and inflammation [117]. Liraglutide reduced hepatitis activity and liver fibrosis as well as attenuated HCB in NASH patients regardless of the presence of T2DM, as determined by the phase 2 LEAN study [118]. A recent meta-analysis by Ghosal et al., including 8 RCT and over 600 patients, found that GLP1R agonists in general improve liver function and histology by improving glycaemia, reducing body weight and hepatic fat content, which in turn might be beneficial for hepatic inflammation in NAFLD concomitant with T2DM [119].
While both GIP and GLP1 are potent insulin secretagogues, GIP has a more robust, dose-dependent secretory pattern, and appears to make a greater contribution than GLP1 to prandial insulin secretion in healthy subjects, while in T2M its activity is depleted. In addition, GIP, but not GLP1, stimulates glucagon secretion by the α-cells at low glycaemia under physiological conditions, and in a glucose-independent fashion in T2M [108]. GIP receptors (GIPR) upregulate lipogenesis, FA esterification and TAG accumulation in adipocytes, and inhibits prandial lipid absorption. Supraphysiological levels of GIP have been associated with increased systemic inflammatory response, and are considered a risk factor for the development of NASH [120].
Tirzepatide (LY3298176) is a synthetic injectable dual GLP1/GIP peptide agonist currently researched for NAFLD treatment. In T2DM subjects with NASH, tirzepatide (1–15 mg/week for 26 weeks) effectively reduced ALT, AST, cytokeratin 18, Pro-C3, and increased adiponectin levels [121]. Additionally, tirzepatide treatment led to greater improvements in liver fat content compared to titrated insulin degludec in T2DM, according to the phase 3 SURPASS-3 RCT results [122]. A phase 2 study, designated SYNERGY-NASH, has been initiated to evaluate the efficacy of tirzepatide in nondiabetic subjects with NASH (NCT04166773).
Oxyntomodulin shares sequence similarity with both and GLP1 and glucagon, and activates GLP1R and glucagon receptors (GCGR) under physiological conditions. Simultaneous GLP1R activation prevents hyperglycaemic response characteristic of glucagon, at the same time potentiating its catabolic effects and greatly intensifying hepatic glycolysis, glycogenolysis, and lipolysis [123]. Weight reduction, anorexigenic and hypoglycaemic effects have been linked to GLP1 activation, while GCGR activation is thought to contribute primarily to hepatic steatosis attenuation and improved mitochondrial respiration. The clinical utility of oxyntomodulin itself is limited by a short circulatory half-life due to rapid renal clearance and degradation by dipeptidyl peptidase 4 (DPP4) [124].
In contrast to the native hormone, synthetic oxyntomodulin mimetics are resistant to proteolytic cleavage and have prolonged pharmacological action. Cotadutide (MEDI0382) (100–300 µg/d) caused substantial improvements in liver enzyme levels and markers of liver fibrosis in concomitant obesity, T2DM, and NASH in a phase 2b study that included over 800 subjects [125]. Efinopegdutide (HM12525A, MK-6024), a PEGylated long-acting peptide agent, has demonstrated promising antihyperlipidaemic, antisteatotic, and anti-inflammatory activity in mice and hamsters [126], and is going to be evaluated in a phase 2 RCT in NASH with semaglutide as an active comparator (NCT04944992). Other dual GLP1/GCCR agonists intended for use in NAFLD include pemvidutide (ALT-801) (NCT05006885), danuglipron (PF-06882961) (in combination with ervogastat) [127], BI 456906 (NCT04771273), and HM14320 (a glucagon-containing combination) [128]. Finally, a novel triple GLP1R/GCGR/GIPR agonist, HM15211, induced significant reductions in liver steatosis, fibrosis, and inflammation in mice [129]; a phase 2 clinical trial is ongoing (NCT04505436).
GLP2, usually not considered an incretin, is prevalent in the gastrointestinal tract, where it promotes lipid absorption, regulates intestinal motility, mucosal morphology, function and integrity of the intestine [130]. Teduglutide, a selective GLP2R agonist, reduced liver steatosis and disease activity scores in rats, possibly by restoring normal intestinal permeability [131].
DPP4 inhibitors represent a group of indirect incretin mimetics as they prevent the proteolytic cleavage of GLP1, GIP, and oxyntomodulin. Threre is no DPP4 inhibitors are yet in the global pipeline for liver disease. However, a number of small-scale clinical trials have evaluated their potential efficacy in NAFLD in the presence or absence of concomitant T2DM. Among this group, only sitagliptin (100 mg/d) was found effective against hepatic steatosis and HCB irrespective of T2DM in a 1-year open-label RCT [132]. Vildagliptin (100 mg/d) [133], saxagliptin (5 mg/d) [134], omarigliptin (25 mg/week) [135], and teneligliptin (20 mg/d) [136] improved liver function and some non-invasive markers of NAFLD, and alogliptin (25 mg/d) was only moderately effective against NASH over 12 months of treatment in T2DM/NAFLD patients [137]. A recent meta-analysis by dos Santos et al. found the existing evidence for DPP4 inhibitors in NAFLD to be of poor quality and altogether not supportive of their clinical effectiveness [138]. Evogliptin [139], anagliptin [140][141], trelagliptin [142], gemigliptin [143], and linagliptin [144] have demonstrated beneficial effects in experimental rodent models, but their clinical value remains to be explored in future trials.
6.4. SGLT Inhibitors
Sodium/glucose cotransporter (SGLT) 2 inhibitors are a relatively novel class of oral antidiabetic agents that increase urinary glucose excretion by inhibiting glucose reabsorption by SGLT2 in the proximal tubules. Several trials have demonstrated the improvement of cardiovascular and renal outcomes by treatment with compounds of this class, namely, empagliflozin, canagliflozin, and dapagliflozin [145]. SGLT2 inhibitors are known for their multiple metabolic effects that are notably relevant to NAFLD pathophysiology, including the general shift towards increased ketogenesis, gluconeogenesis, glycogenolysis, and FA β-oxidation. They inhibit leptin production by adipocytes, leading to decreased food intake, increase adiponectin levels, provide mild insulin sensitization, suppress HSC activation and fibrogenesis. Additionally, SGLT2 inhibitors may indirectly suppress sympathetic innervation and increase the vagal tone, thereby preventing the activation of Kupffer cells and the associated inflammatory processes [146][147].
Recent evidence mostly supports the efficacy of the majority of SGLT2 inhibitors for improving liver dysfunction, steatosis and fibrosis in NAFLD concomitant with T2DM. Among this group, only dapagliflozin, empagliflozin, and canagliflozin treatment was associated with beneficial effects in nondiabetic NAFLD patients. Dapagliflozin (10 mg) significantly reduced ALT, AST, and GGT levels, according to a retrospective study [148], while empagliflozin (10 mg/d) also attenuated liver steatosis and liver stiffness, indicative of potential antifibrotic activity, in a small-scale RCT [149]. The phase 3 DEAN trial to evaluate dapagliflozin in biopsy-confirmed NASH patients has been initiated (NCT03723252). Canagliflozin (100 mg/d) improved liver enzyme levels and FIB-4 index values in an open-label, uncontrolled pilot study [150]. Additionally, empagliflozin had a beneficial effect on cognitive functions and reduced anxiety in an experimental NAFLD model [151].
Ipragliflozin (50 mg/d for 72 weeks) ameliorated liver fibrosis and enhanced NASH resolution [152], remogliflozin etabonate (50–1000 mg/d for 12 weeks) reduced FIB-4 and NAFLD-fibrosis scores [153], and ertugliflozin (5 or 15 mg/d for 52 weeks) reduced liver transaminase levels [156] in TD2M patients with different stages of NASH. Several pilot studies in T2DM/NAFLD subjects confirmed the antisteatotic properties of luseogliflozin and tofogliflozin [155][156][157].
The SGLT1 subtype plays a relatively smaller (10–20% and up to 40% when SGLT2 are blocked) role in the renal glucose reabsorption, but is more abundant in the small intestine along with the heart and lungs. Intestinal SGLT1 (iSGLT1) inhibition leads to substantially reduced glucose and galactose absorption from the intestinal lumen, and increased incretin (GLP1, peptide YY) release by enteroendocrine cells [158]. Currently, licogliflozin (LIK066) is the only SGLT1/2 inhibitor being evaluated in NASH independent of T2DM presence, alone and in combination with the FXR agonist tropifexor, in the ongoing phase 2 ELIVATE study (NCT04065841). Previously, licogliflozin (150 mg/d) reduced ALT, ALT, GGT levels and liver fat content over 12 weeks compared to placebo [159]. A novel compound, SGL5213, has been identified as a selective iSGLT1 inhibitor, and has demonstrated insulin-sensitizing, anti-inflammatory, and antifibrotic activity in a murine model of NAFLD [160].
6.5. a-Glucosidase Inhibitors
α-Glucosidase is a carbohydrate hydrolase located in the brush border of the small intestine that catalyzes the breakdown of dietary starch and disaccharides to yield glucose. α-Glucosidase inhibitors slow down carbohydrate digestion and absorption, thereby reducing postprandial hyperglycaemia. However, they are characterized by only modest overall antidiabetic activity, and are not too often used in clinical practice [161]. Despite some scientific interest concerning the use of this class of drugs for the treatment of liver diseases, data regarding their efficacy for NAFLD remain scarce. Acarbose (100 mg/d) improved AST, ALT levels and lipid profiles, albeit to a lesser extent than ezetimibe, in a 10-week small-scale RCT in non-diabetic NASH patients [162]. Miglitol treatment (150 mg/d for 12 months) was associated with significant improvements in steatosis, lobular and portal inflammation, and NAS scores, while fibrosis and hepatocyte ballooning remained unchanged [163]. Finally, voglibose prevented hepatic steatosis in obese rats, but was slightly inferior to empagliflozin [164].
7. Other Agents
7.1. Probiotics
7.2. Mesenchymal Stromal Cells
Lately, cell-based therapy has emerged as a feasible alternative for the treatment of different stages of NAFLD. In particular, experimental evidence supports the use of bone marrow- [176], umbilical cord- [177], and compact bone-derived mesenchymal stromal cells [178] as well as hepatocytes derived by differentiating induced pluripotent stem cells [179]. The crosstalk between hepatic stem cells and their possible therapeutic application for NAFLD are discussed in detail in a recent review by Overi et al. [180].
HepaStem® is a first-in-class allogeneic stem cell therapy product containing human adult liver-derived progenitor cells with potential indications including cirrhotic and precirrhotic NASH as well as acute-on-chronic liver failure (ACLF). HepaStem® cells, obtained from healthy donors, are expected to modulate the inflammatory response and inhibit HSC activation, thereby reducing liver fibrosis. A small-scale phase 2a RCT found HepaStem® to be safe and well tolerated, and indicated potential efficacy for ACLF and/or decompensated liver cirrhosis [181].
7.3. Fraudulent Fatty Acids
Fraudulent, or abnormal fatty acids, represented by bempedoic acid (ETC-1002) and gemcabene (CI-1027), are molecules with structures similar to those of oleic or linolenic acid that regulate metabolic pathways in the liver, resulting in enhanced FA catabolism. After conversion into its active form, bempedoic acid acts as false substrate and inhibits hepatic adenosine triphosphate citrate (pro-S)-lyase, an enzyme upstream of 3-hydroxy-3-methylglutaryl-CoA reductase in the cholesterol synthesis pathway. This links fraudulent fatty acids to statins, whose possible beneficial effects for NAFLD are reviewed elsewhere [182].
Bempedoic acid is approved in the USA and EU as monotherapy and as a fixed dose combination with ezetimibe for the treatment of hypercholesterolaemia [183]. In a high-fat diet-induced murine model of NASH, it caused significant reductions in ALT and AST levels, hepatic TAG accumulation, proinflammatory and profibrotic gene expression, resulting in improved NAFLD activity and liver fibrosis by histological analysis [184].
Gemcabene (PD-72953), a structurally optimized derivative of bempedoic acid, forms a CoA conjugate that inhibits ACC, and reduces apolipoprotein C-III expression [183]. In a mouse model of NASH/HCC, it diminished micro- and macrovesicular liver steatosis, HCB, inflammatory infiltration, and fibrosis, which corresponded to downregulated proinflammatory, lipogenesis, and profibrogenic marker expression [183]. Gemcabene was being developed for the treatment of paediatric NAFLD, but was discontinued and repurposed for another indication after a lack of efficacy was demonstrated in a phase 2a proof-of-concept study (NCT03436420).
7.4. Tesamorelin
Tesamorelin (TH9507) is a growth hormone (GH) releasing hormone analogue that is thought to stimulate lipolysis via increasing endogenous GH levels while maintaining feedback inhibition and limiting toxicity compared to native GH. Tesamorelin reduced liver fat content and visceral fat in a preliminary study in antiretroviral-treated patients with human immunodeficiency virus (HIV)-associated lipodystrophy [185]. A phase 2 trial to evaluate the effects of tesamorelin on liver steatosis and cardiovascular risk in obese NASH
patients is recruiting (NCT03375788), and a phase 3 study in the general population with NAFLD including a HIV cohort has been planned [186].
7.5. Berberine Ursodeoxycholate
Berberine ursodeoxycholate (BUDCA, HTD1801) is an ionic salt of the isoquinoline alkaloid berberine and ursodeoxycholic acid (UDCA). According to a meta-analysis by Wei et al., berberine can significantly improve liver function, lipid profiles, and glycaemic control in patients with NAFLD [187] due to adenosine monophosphate-activated protein kinase (AMPK) activation, stimulation of glycolysis, and, possibly, inhibition of α-glucosidase [188]. UDCA, in turn, is a bile acid long used for the treatment of NASH and chronic cholestatic diseases, whose hepatoprotective effects are confirmed by several systematic reviews and meta-analyses [189][190]. In a phase 2 proof-of-concept RCT in T2DM patients with presumed NASH, BUDCA reduced liver enzyme levels and liver steatosis by MRI-PDFF [191].
7.6. Miricorilant
Miricorilant (CORT 118335) is an investigational glucocorticoid receptor agonist/antagonist and a mineralocorticoid receptor antagonist currently in development for NASH and antipsychotic-induced weight gain. Results of a phase 2a study in NASH patients demonstrated that miricorilant (600 mg/d) effectively ameliorated liver steatosis by radiological measures. However, miricorilant treatment was associated by transient yet significant increases in serum transaminases [192], and the trial was subsequently put on hold due to safety concerns (NCT03823703).
7.7. Nitazoxanide
Nitazoxanide (Alinia®) is an FDA-approved broad-spectrum thiazolide antiprotozoal and antiparasitic agent, lately reported to be a potent AMPK activator and inhibitor of HSC activation. In experimental studies in mice, nitazoxanide (100 mg/kg/d) attenuated dyslipidaemia, liver steatosis [193], fibrosis, inflammation, and HCB, demonstrating synergistic effects with the pan-PPAR agonist elafibranor [194]. Moreover, the anti-anaerobic activity of nitazoxanide may determine its use in preventing the recurrence of hepatic encephalopathy as a viable alternative to rifaximin (NCT04161053) [195].
7.8. Pirfenidone
Pirfenidone (Esbriet®) is a pyridine derivative with antifibrotic, anti-inflammatory, and antioxidant properties, the precise mechanisms of which are still unclear. In the liver, pirfenidone may decrease fibronectin, TGFβ, collagen production and attenuate fibrogenesis, hepatocyte necrosis, and necroinflammation [196][197]. In the phase 2 PROMETEO study, pirfenidone (1200 mg/d) markedly reduced transaminase levels and advanced liver fibrosis of predominantly nonalcoholic aetiology [198].
7.9. Miscellaneous
The modern pipeline for NAFLD includes a plethora of drug candidates with diverse and innovative mechanisms of action. Other investigational drugs with potential therapeutic value in NAFLD include antileukotriene agents [199], GPCR modulators [200], anti-IL mABs [201], IL22 axis modulators [202], purinergic receptor agonists [203], antioxidants [204], antisense oligonucleotides [205], multitarget epigenetic regulators [206], and many more. An overview of the current drug development pipeline for NAFLD is given in Figure 1. Given the exceptionally complex pathophysiology and the multifaceted nature of this disease, NAFLD pharmacotherapy can be expected to remain a priority for biomedical research in the nearest future.
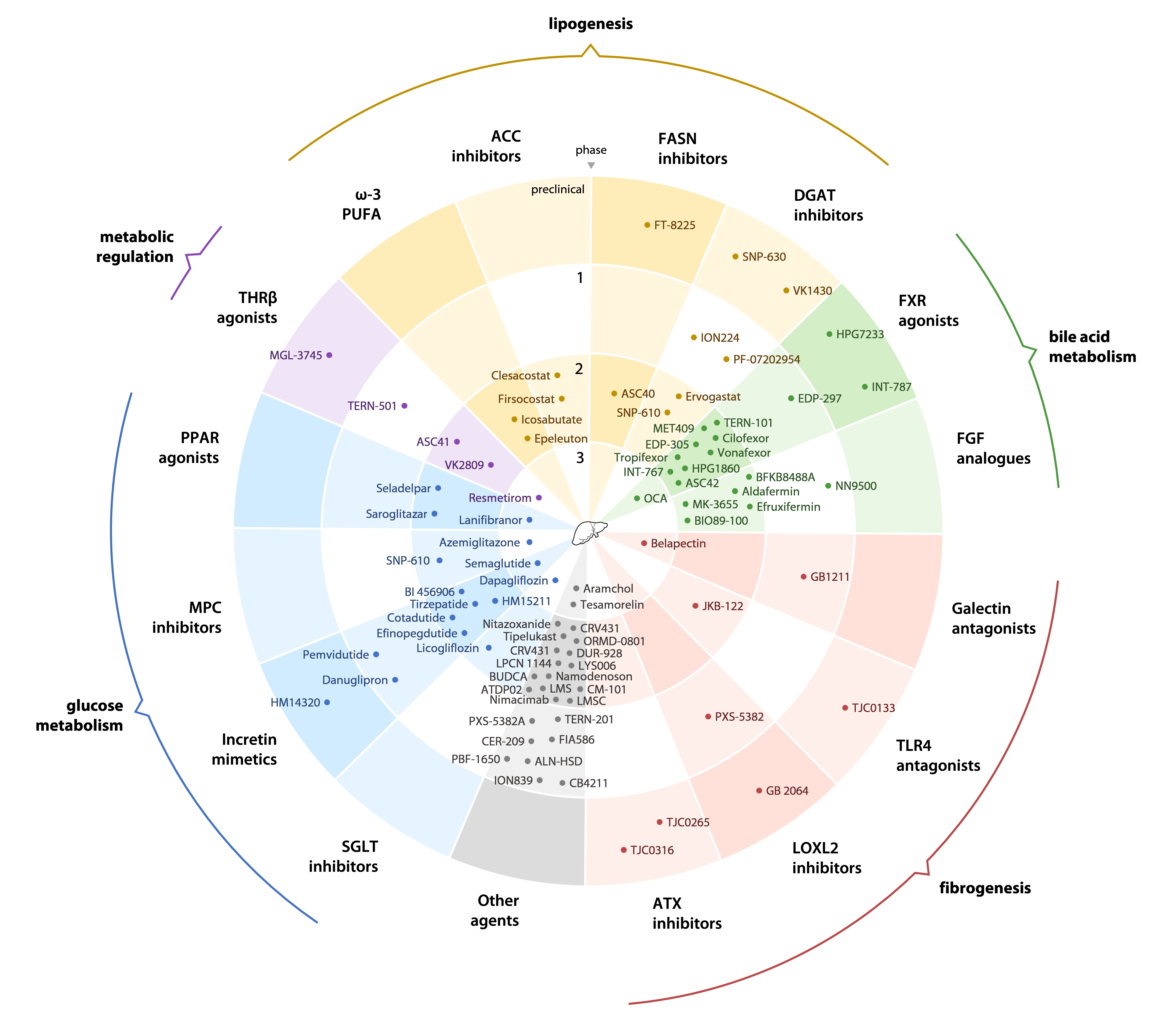 Figure 1. An overview of the current drug development pipeline for non-alcoholic fatty liver disease.
Figure 1. An overview of the current drug development pipeline for non-alcoholic fatty liver disease.
FASN, fatty acid synthase; DGAT, diglyceride acyltransferase; FXR, farnesoid X receptor; FGF, fibroblast growth factor; TLR4, toll-like receptor 4; LOXL2, lysyl oxidase-like protein 2; ATX, autotaxin; SGLT, sodium/glucose contransporter; MPC, mitochondrial pyruvate carrier; PPAR, peroxisome proliferator-activated receptor; THRβ, thyroid hormone receptor β; PUFA, polyunsaturated fatty acid; ACC, acetyl-CoA carboxylase; BUDCA, berberine ursodeoxycholate; LMS, leucine-metforminsildenafil; LMSC, liver-derived mesenchymal stromal cells.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10020274
References
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402.
- Monelli, F.; Venturelli, F.; Bonilauri, L.; Manicardi, E.; Manicardi, V.; Giorgi Rossi, P.; Massari, M.; Ligabue, G.; Riva, N.; Schianchi, S.; et al. Systematic review of existing guidelines for NAFLD assessment. Hepatoma Res. 2021, 7, 25.
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133.
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1.
- Ortiga-Carvalho, T.M.; Sidhaye, A.R.; Wondisford, F.E. Thyroid hormone receptors and resistance to thyroid hormone disorders. Nat. Rev. Endocrinol. 2014, 10, 582–591.
- Pramfalk, C.; Pedrelli, M.; Parini, P. Role of thyroid receptor β in lipid metabolism. Biochim. Biophys. Acta 2011, 1812, 929–937.
- Dawson, P.A.; Parini, P. Hepatic thyroid hormone receptor β1 agonism: Good for lipids, good for bile? J. Lipid Res. 2018, 59, 1551–1553.
- Gionfra, F.; De Vito, P.; Pallottini, V.; Lin, H.Y.; Davis, P.J.; Pedersen, J.Z.; Incerpi, S. The Role of Thyroid Hormones in Hepatocyte Proliferation and Liver Cancer. Front. Endocrinol. 2019, 10, 532.
- Harrison, S.A.; Bashir, M.; Moussa, S.E.; McCarty, K.; Frias, J.P.; Taub, R.; Alkhouri, N. Effects of Resmetirom on Noninvasive Endpoints in a 36-Week Phase 2 Active Treatment Extension Study in Patients with NASH. Hepatol. Commun. 2021, 5, 573–588.
- Loomba, R.; Neutel, J.; Mohseni, R.; Bernard, D.; Severance, R.; Dao, M.; Saini, S.; Margaritescu, C.; Homer, K.; Tran, B.; et al. VK2809, a Novel Liver-Directed Thyroid Receptor Beta Agonist, Significantly Reduces Liver Fat with Both Low and High Doses in Patients with Non-Alcoholic Fatty Liver Disease: A Phase 2 Randomized, Placebo-Controlled Trial. J. Hepatol. 2019, 70, e141–e382.
- Li, X.; He, H.; Sho, E.; Yang, B.; Wu, J.J. Significant Improvement of NAFLD Activity Scores and Liver Fibrosis by ASC41, a Selective THR-β Agonist, in High Fat Diet Induced NASH SD Rats. In Proceedings of the Digital International Liver Congress 2021, Virtual, 23–26 June 2021.
- Wu, J.; He, H.; Palmer, M.; Yan, Y. A Phase Ib Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Asc41, A THR-Β Agonist, for 28-Days in Overweight and Obese Subjects with Elevated LDL-C, a Population Characteristic of NAFLD. In Proceedings of the Liver Meeting 2021, Virtual, 12–15 November 2021.
- Kirschberg, T.; Jones, C.; Xu, Y.; Wang, Y.; Fenaux, M.; Klucher, K. TERN-501, a Potent and Selective Agonist of Thyroid Hormone Receptor Beta, Strongly Reduces Histological Features and Biomarkers of Non-Alcoholic Steatohepatitis Associated Pathology in Rodent Models. In Proceedings of the Digital International Liver Congress 2020, Virtual, 27–29 August 2020.
- Ascletis Pharma Inc. Available online: https://ascletis.com/ (accessed on 10 December 2021).
- Terns Pharmaceuticals. Available online: https://www.ternspharma.com/ (accessed on 10 December 2021).
- Qi, J.; Lang, W.; Geisler, J.G.; Wang, P.; Petrounia, I.; Mai, S.; Smith, C.; Askari, H.; Struble, G.T.; Williams, R.; et al. The use of stable isotope-labeled glycerol and oleic acid to differentiate the hepatic functions of DGAT1 and -2. J. Lipid Res. 2012, 53, 1106–1116.
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374.
- Takemoto, K.; Fukasaka, Y.; Yoshimoto, R.; Nambu, H.; Yukioka, H. Diacylglycerol acyltransferase 1/2 inhibition induces dysregulation of fatty acid metabolism and leads to intestinal barrier failure and diarrhea in mice. Physiol. Rep. 2020, 8, e14542.
- Parlati, L.; Régnier, M.; Guillou, H.; Postic, C. New targets for NAFLD. JHEP Rep. 2021, 3, 100346.
- Panzitt, K.; Wagner, M. FXR in liver physiology: Multiple faces to regulate liver metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166133.
- Loomba, R.; Noureddin, M.; Kowdley, K.V.; Kohli, A.; Sheikh, A.; Neff, G.; Bhandari, B.R.; Gunn, N.; Caldwell, S.H.; Goodman, Z.; et al. Combination Therapies Including Cilofexor and Firsocostat for Bridging Fibrosis and Cirrhosis Attributable to NASH. Hepatology 2021, 73, 625–643.
- Calle, R.A.; Amin, N.B.; Carvajal-Gonzalez, S.; Ross, T.T.; Bergman, A.; Aggarwal, S.; Crowley, C.; Rinaldi, A.; Mancuso, J.; Aggarwal, N.; et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: Two parallel, placebo-controlled, randomized phase 2a trials. Nat. Med. 2021, 27, 1836–1848.
- Syed-Abdul, M.M.; Parks, E.J.; Gaballah, A.H.; Bingham, K.; Hammoud, G.M.; Kemble, G.; Buckley, D.; McCulloch, W.; Manrique-Acevedo, C. Fatty Acid Synthase Inhibitor TVB-2640 Reduces Hepatic de Novo Lipogenesis in Males with Metabolic Abnormalities. Hepatology 2020, 72, 103–118.
- Loomba, R.; Rinella, M.; Harrison, S.A.; Wong, V.W.-S.; Ratziu, V.; Mohseni, R.; Lucas, J.; Gutierrez, J.A.; Rahimi, R.; Trotter, J.; et al. Novel, first-in-class, fatty acid synthase inhibitor, TVB-2640 versus placebo demonstrates clinically significant reduction in liver fat by MRI-PDFF in NASH. In Proceedings of the Digital International Liver Congress 2020, Virtual, 27–29 August 2020.
- Forma Therapeutics. Available online: https://www.formatherapeutics.com/ (accessed on 10 December 2021).
- Bhattacharya, D.; Basta, B.; Mato, J.M.; Craig, A.; Fernández-Ramos, D.; Lopitz-Otsoa, F.; Tsvirkun, D.; Hayardeny, L.; Chandar, V.; Schwartz, R.E.; et al. Aramchol downregulates stearoyl CoA-desaturase 1 in hepatic stellate cells to attenuate cellular fibrogenesis. JHEP Rep. 2021, 3, 100237.
- Leikin-Frenkel, A.; Gonen, A.; Shaish, A.; Goldiner, I.; Leikin-Gobbi, D.; Konikoff, F.M.; Harats, D.; Gilat, T. Fatty acid bile acid conjugate inhibits hepatic stearoyl coenzyme A desaturase and is non-atherogenic. Arch. Med. Res. 2010, 41, 397–404.
- Ratziu, V.; de Guevara, L.; Safadi, R.; Poordad, F.; Fuster, F.; Flores-Figueroa, J.; Arrese, M.; Fracanzani, A.L.; Ben Bashat, D.; Lackner, K.; et al. Aramchol in patients with nonalcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase 2b trial. Nat. Med. 2021, 27, 1825–1835.
- Calle, R.; Aggarwal, S.; Mancuso, J.; Bergman, A.; Somayaji, V.; Ross, T.; Esler, W. Co-administration of PF-05221304 and PF-06865571 delivers robust whole liver fat reduction and mitigation of acetyl-coa carboxilase inhibitor induced hypertriglyceridemia in patients with NAFLD. J. Hepatol. 2020, 73, S401–S652.
- Loomba, R.; Morgan, E.; Watts, L.; Xia, S.; Hannan, L.A.; Geary, R.S.; Baker, B.F.; Bhanot, S. Novel antisense inhibition of diacylglycerol O-acyltransferase 2 for treatment of non-alcoholic fatty liver disease: A multicentre, double-blind, randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol. Hepatol. 2020, 5, 829–838.
- Sinew Pharma Inc. Available online: https://www.sinewpharma.com/en/ (accessed on 10 December 2021).
- Dentin, R.; Benhamed, F.; Pégorier, J.P.; Foufelle, F.; Viollet, B.; Vaulont, S.; Girard, J.; Postic, C. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J. Clin. Investig. 2005, 115, 2843–2854.
- Calder, P.C. n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am. J. Clin. Nutr. 2006, 83, 1505S–1519S.
- Siriwardhana, N.; Kalupahana, N.S.; Moustaid-Moussa, N. Health benefits of n-3 polyunsaturated fatty acids: Eicosapentaenoic acid and docosahexaenoic acid. Adv. Food Nutr. Res. 2012, 65, 211–222.
- Okada, L.S.D.R.R.; Oliveira, C.P.; Stefano, J.T.; Nogueira, M.A.; Silva, I.D.C.G.D.; Cordeiro, F.B.; Alves, V.A.F.; Torrinhas, R.S.; Carrilho, F.J.; Puri, P.; et al. Omega-3 PUFA modulate lipogenesis, ER stress, and mitochondrial dysfunction markers in NASH-Proteomic and lipidomic insight. Clin. Nutr. 2018, 37, 1474–1484.
- Nogueira, M.A.; Oliveira, C.P.; Ferreira Alves, V.A.; Stefano, J.T.; Rodrigues, L.S.; Torrinhas, R.S.; Cogliati, B.; Barbeiro, H.; Carrilho, F.J.; Waitzberg, D.L. Omega-3 polyunsaturated fatty acids in treating non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled trial. Clin. Nutr. 2016, 35, 578–586.
- Parker, H.M.; Johnson, N.A.; Burdon, C.A.; Cohn, J.S.; O’Connor, H.T.; George, J. Omega-3 supplementation and non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2012, 56, 944–951.
- Nobili, V.; Bedogni, G.; Alisi, A.; Pietrobattista, A.; Risé, P.; Galli, C.; Agostoni, C. Docosahexaenoic acid supplementation decreases liver fat content in children with non-alcoholic fatty liver disease: Double-blind randomised controlled clinical trial. Arch. Dis. Child. 2011, 96, 350–353.
- He, X.X.; Wu, X.L.; Chen, R.P.; Chen, C.; Liu, X.G.; Wu, B.J.; Huang, Z.M. Effectiveness of Omega-3 Polyunsaturated Fatty Acids in Non-Alcoholic Fatty Liver Disease: A Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2016, 11, e0162368.
- Yan, J.H.; Guan, B.J.; Gao, H.Y.; Peng, X.E. Omega-3 polyunsaturated fatty acid supplementation and non-alcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Medicine 2018, 97, e12271.
- Lee, C.H.; Fu, Y.; Yang, S.J.; Chi, C.C. Effects of Omega-3 Polyunsaturated Fatty Acid Supplementation on Non-Alcoholic Fatty Liver: A Systematic Review and Meta-Analysis. Nutrients 2020, 12, 2769.
- Climax, J.; Newsome, P.N.; Hamza, M.; Weissbach, M.; Coughlan, D.; Sattar, N.; McGuire, D.K.; Bhatt, D.L. Effects of Epeleuton, a Novel Synthetic Second-Generation n-3 Fatty Acid, on Non-Alcoholic Fatty Liver Disease, Triglycerides, Glycemic Control, and Cardiometabolic and Inflammatory Markers. J. Am. Heart Assoc. 2020, 9, e016334.
- Affimune. Available online: https://www.afimmune.com/ (accessed on 10 December 2021).
- Harrison, S.; Gunn, N.T.; Sheikh, M.Y.; Rudraraju, M.; Kohli, A.; Neff, G.; Round, P.; Fraser, D.A.; Beysen, C.; Rossi, S.; et al. Icosabutate, a novel structurally engineered fatty acid, significantly reduces relevant markers of NASH and fibrosis in 16 weeks: Interim analysis results of the ICONA trial. J. Hepatol. 2021, 75, S294–S803.
- Jiang, L.; Zhang, H.; Xiao, D.; Wei, H.; Chen, Y. Farnesoid X receptor (FXR): Structures and ligands. Comput. Struct. Biotechnol. J. 2021, 19, 2148–2159.
- Intercept Pharmaceuticals. Available online: https://www.interceptpharma.com/homepage-non-usa/ (accessed on 10 December 2021).
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582.e1.
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965.
- Alemi, F.; Kwon, E.; Poole, D.P.; Lieu, T.; Lyo, V.; Cattaruzza, F.; Cevikbas, F.; Steinhoff, M.; Nassini, R.; Materazzi, S.; et al. The TGR5 receptor mediates bile acid-induced itch and analgesia. J. Clin. Investig. 2013, 123, 1513–1530.
- An, P.; Wei, G.; Huang, P.; Li, W.; Qi, X.; Lin, Y.; Vaid, K.A.; Wang, J.; Zhang, S.; Li, Y.; et al. A novel non-bile acid FXR agonist EDP-305 potently suppresses liver injury and fibrosis without worsening of ductular reaction. Liver Int. 2020, 40, 1655–1669.
- Chau, M.; Li, Y.; Roqueta-Rivera, M.; Garlick, K.; Shen, R.; Wang, G.; Or, Y.S.; Jiang, L.-J. Characterization of EDP-305, a Highly Potent and Selective Farnesoid X Receptor Agonist, for the Treatment of Non-alcoholic Steatohepatitis. Int. J. Gastroenterol. 2019, 3, 4–16.
- Enanta Pharmaceuticals, Inc. Enanta Announces Results of INTREPID Study of EDP-305 for the Treatment of Primary Biliary Cholangitis: News Release; 2020. Available online: https://www.enanta.com/investors/news-releases/press-release/2020/Enanta-Announces-Results-of-INTREPID-Study-of-EDP-305-for-the-Treatment-of-Primary-Biliary-Cholangitis/default.aspx (accessed on 10 December 2021).
- Biagioli, M.; Fiorucci, S. Bile acid activated receptors: Integrating immune and metabolic regulation in non-alcoholic fatty liver disease. Liver Res. 2021, 5, 119–141.
- Enanta Pharmaceuticals, Inc. Enanta Pharmaceuticals Provides Update on NASH FXR Agonist Programs: News Release; 2021. Available online: https://www.enanta.com/investors/news-releases/press-release/2021/Enanta-Pharmaceuticals-Provides-Update-on-NASH-FXR-Agonist-Programs/ (accessed on 10 December 2021).
- Carino, A.; Cipriani, S.; Marchianò, S.; Biagioli, M.; Santorelli, C.; Donini, A.; Zampella, A.; Monti, M.C.; Fiorucci, S. BAR502, a dual FXR and GPBAR1 agonist, promotes browning of white adipose tissue and reverses liver steatosis and fibrosis. Sci. Rep. 2017, 7, 42801.
- Roth, J.D.; Feigh, M.; Veidal, S.S.; Fensholdt, L.K.; Rigbolt, K.T.; Hansen, H.H.; Chen, L.C.; Petitjean, M.; Friley, W.; Vrang, N.; et al. INT-767 improves histopathological features in a diet-induced ob/ob mouse model of biopsy-confirmed non-alcoholic steatohepatitis. World J. Gastroenterol. 2018, 24, 195–210.
- Harrison, S.A.; Bashir, M.R.; Lee, K.J.; Shim-Lopez, J.; Lee, J.; Wagner, B.; Smith, N.D.; Chen, H.C.; Lawitz, E.J. A structurally optimized FXR agonist, MET409, reduced liver fat content over 12 weeks in patients with non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 25–33.
- Kremoser, C. FXR agonists for NASH: How are they different and what difference do they make? J. Hepatol. 2021, 75, 12–15.
- Schumacher, J.D.; Guo, G.L. Pharmacologic Modulation of Bile Acid-FXR-FGF15/FGF19 Pathway for the Treatment of Nonalcoholic Steatohepatitis. Handb. Exp. Pharmacol. 2019, 256, 325–357.
- Lucas, K.J.; Lopez, P.; Lawitz, E.; Sheikh, A.; Aizenberg, D.; Hsia, S.; Boon Bee, G.G.; Vierling, J.; Frias, J.; White, J.; et al. Tropifexor, a highly potent FXR agonist, produces robust and dose-dependent reductions in hepatic fat and serum alanine aminotransferase in patients with fibrotic NASH after 12 weeks of therapy: FLIGHT-FXR Part C interim results. Dig. Liver Dis. 2020, 52, e19–e45.
- Novartis. A Randomized, Double-Blind, Placebo Controlled, 3-Part, Adaptive Design, Multicenter Study to Assess Safety, Tolerability and Efficacy of Tropifexor (LJN452) in Patients with Non-Alcoholic Steatohepatitis (NASH): FLIGHT-FXR, 2020. Available online: https://www.novctrd.com/ctrdweb/trialresult/trialresults/pdf?trialResultId=17816 (accessed on 10 December 2021).
- Patel, K.; Harrison, S.A.; Elkhashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.E.; Kayali, Z.; Wong, V.W.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients with Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71.
- Harrison, S.A.; Ratziu, V.; White, A.; Reiss, G.M.; Bowman, W.K.; Gunn, N.; Loustaud Ratti, V.; Bureau, C.; Lawitz, E.J.; Alkhouri, N.; et al. Vonafexor, a FXR Agonist, Induced Hepatic and Renal Improvement in the Randomized, Double-Blind, Placebo controlled LIVIFYNASH Trial. In Proceedings of the Liver Meeting 2021, Virtual, 12–15 November 2021.
- Novartis. A randomized, Patient and Investigator Blinded, Placebo-Controlled, Multicenter Study to Assess the Safety, Tolerability, Pharmacokinetics and Efficacy of LMB763 in Patients with Non-Alcoholic Steatohepatitis (NASH), 2020. Available online: https://www.novctrd.com/ctrdweb/trialresult/trialresults/pdf?trialResultId=17527 (accessed on 10 December 2021).
- Loomba, R.; Kowdley, K.V.; Vuppalanchi, R.; Hassanein, T.; Rojter, S.E.; Sheikh, M.Y.; Moussa, S.; Chung, D.; Eng, C.; Marmon, T.; et al. Liver-Distributed FXR Agonist TERN-101 Demonstrates Favorable Safety and Efficacy Profile in NASH Phase 2a LIFT Study. Hepatology 2021, 74, 97A–98A.
- Hepagene Therapeutics, Inc. Available online: http://www.hepagene.com/#/m_about2 (accessed on 10 December 2021).
- Henriksson, E.; Andersen, B. FGF19 and FGF21 for the Treatment of NASH-Two Sides of the Same Coin? Differential and Overlapping Effects of FGF19 and FGF21 from Mice to Human. Front. Endocrinol. 2020, 11, 601349.
- Harrison, S.A.; Abdelmalek, M.F.; Neff, G.W.; Gunn, N.; Guy, C.D.; Alkhouri, N.; Bashir, M.; Freilich, B.; Almeda, J.; Knapple, W.; et al. Topline Results from the ALPINE 2/3 Study: A Randomized, Double-Blind, Placebo-Controlled, Multicenter, Phase 2b Trial Evaluating 3 Doses of the FGF19 Analogue Aldafermin on Liver Histology in Patients with Nonalcoholic Steatohepatitis and Stage 2 or 3 Fibrosis. Hepatology 2021, 74, 5A.
- Harrison, S.A.; Ruane, P.J.; Freilich, B.L.; Neff, G.; Patil, R.; Behling, C.A.; Hu, C.; Fong, E.; de Temple, B.; Tillman, E.J.; et al. Efruxifermin in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled, phase 2a trial. Nat. Med. 2021, 27, 1262–1271.
- Frias, J.P.; Lawitz, E.J.; Ortiz-LaSanta, G.; Franey, B.; Morrow, L.; Chen, C.-Y.; Tseng, L.; Charlton, R.W.; Mansbach, H.; Margalit, M.; et al. BIO89-100 Demonstrated Robust Reductions in Liver Fat and Liver Fat Volume (LFV) by MRI-PDFF, Favorable Tolerability and Potential for Weekly (QW) or Every 2 Weeks (Q2W) Dosing in a Phase 1b/2a Placebo-Controlled, Double-Blind, Multiple Ascending Dose Study in NASH. J. Endocr. Soc. 2021, 5, A5–A6.
- Novo Nordisk. Available online: https://www.novonordisk.com/ (accessed on 10 December 2021).
- Baruch, A.; Wong, C.; Chinn, L.W.; Vaze, A.; Sonoda, J.; Gelzleichter, T.; Chen, S.; Lewin-Koh, N.; Morrow, L.; Dheerendra, S.; et al. Antibody-mediated activation of the FGFR1/Klothoβ complex corrects metabolic dysfunction and alters food preference in obese humans. Proc. Natl. Acad. Sci. USA 2020, 117, 28992–29000.
- Pan, Q.; Lin, S.; Li, Y.; Liu, L.; Li, X.; Gao, X.; Yan, J.; Gu, B.; Chen, X.; Li, W.; et al. A novel GLP-1 and FGF21 dual agonist has therapeutic potential for diabetes and non-alcoholic steatohepatitis. EBioMedicine 2021, 63, 103202.
- Abdelmalek, M.F.; Sanyal, A.J.; Nakajima, A.; Neuschwander-Tetri, B.A.; Goodman, Z.; Lawitz, E.J.; Harrison, S.A.; Jacobson, I.M.; Imajo, K.; Gunn, N.; et al. Efficacy and Safety of Pegbelfermin in Patients with Nonalcoholic Steatohepatitis and Compensated Cirrhosis: Results from the Phase 2b, Randomized, Double-Blind, Placebo-Controlled Falcon 2 Study. In Proceedings of the Liver Meeting 2021, Virtual, 12–15 November 2021.
- Sun, M.J.; Cao, Z.Q.; Leng, P. The roles of galectins in hepatic diseases. J. Mol. Histol. 2020, 51, 473–484.
- An, Y.; Xu, S.; Liu, Y.; Xu, X.; Philips, C.A.; Chen, J.; Méndez-Sánchez, N.; Guo, X.; Qi, X. Role of Galectins in the Liver Diseases: A Systematic Review and Meta-Analysis. Front. Med. 2021, 8, 744518.
- Traber, P.G.; Chou, H.; Zomer, E.; Hong, F.; Klyosov, A.; Fiel, M.I.; Friedman, S.L. Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide-induced liver disease. PLoS ONE 2013, 8, e75361.
- Chalasani, N.; Abdelmalek, M.F.; Garcia-Tsao, G.; Vuppalanchi, R.; Alkhouri, N.; Rinella, M.; Noureddin, M.; Pyko, M.; Shiffman, M.; Sanyal, A.; et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients with Nonalcoholic Steatohepatitis with Cirrhosis and Portal Hypertension. Gastroenterology 2020, 158, 1334–1345.e5.
- Hsieh, Y.C.; Lee, K.C.; Wu, P.S.; Huo, T.I.; Huang, Y.H.; Hou, M.C.; Lin, H.C. Eritoran Attenuates Hepatic Inflammation and Fibrosis in Mice with Chronic Liver Injury. Cells 2021, 10, 1562.
- Yang, L.; Seki, E. Toll-like receptors in liver fibrosis: Cellular crosstalk and mechanisms. Front. Physiol. 2012, 3, 138.
- Huang, K.C.; Chen, M.Y.; Lin, C.L.; Yang, S.S.; Hsu, C.W.; Wang, C.C.; Huang, Y.H.; Chang, C.C.; Shih, Y.C.; Liu, S.H. JKB-111 in Patients with Non-Alcoholic Fatty Liver: A Phase 2 Randomized Double-Blind Placebo-Control Study. In Proceedings of the Digital International Liver Congress 2020, Virtual, 27–29 August 2020.
- Klepfish, M.; Gross, T.; Vugman, M.; Afratis, N.A.; Havusha-Laufer, S.; Brazowski, E.; Solomonov, I.; Varol, C.; Sagi, I. LOXL2 Inhibition Paves the Way for Macrophage-Mediated Collagen Degradation in Liver Fibrosis. Front. Immunol. 2020, 11, 480.
- Chen, W.; Yang, A.; Jia, J.; Popov, Y.V.; Schuppan, D.; You, H. Lysyl Oxidase (LOX) Family Members: Rationale and Their Potential as Therapeutic Targets for Liver Fibrosis. Hepatology 2020, 72, 729–741.
- Harrison, S.A.; Abdelmalek, M.F.; Caldwell, S.; Shiffman, M.L.; Diehl, A.M.; Ghalib, R.; Lawitz, E.J.; Rockey, D.C.; Schall, R.A.; Jia, C.; et al. Simtuzumab Is Ineffective for Patients with Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 1140–1153.
- Schilter, H.; Findlay, A.D.; Perryman, L.; Yow, T.T.; Moses, J.; Zahoor, A.; Turner, C.I.; Deodhar, M.; Foot, J.S.; Zhou, W.; et al. The lysyl oxidase like 2/3 enzymatic inhibitor, PXS-5153A, reduces crosslinks and ameliorates fibrosis. J. Cell. Mol. Med. 2019, 23, 1759–1770.
- Galecto, Inc. Available online: https://galecto.com/ (accessed on 10 December 2021).
- Geraldo, L.H.M.; Spohr, T.C.L.S.; Amaral, R.F.D.; Fonseca, A.C.C.D.; Garcia, C.; Mendes, F.A.; Freitas, C.; dos Santos, M.F.; Lima, F.R.S. Role of lysophosphatidic acid and its receptors in health and disease: Novel therapeutic strategies. Signal Transduct. Target. Ther. 2021, 6, 45.
- Bain, G.; Shannon, K.E.; Huang, F.; Darlington, J.; Goulet, L.; Prodanovich, P.; Ma, G.L.; Santini, A.M.; Stein, A.J.; Lonergan, D.; et al. Selective Inhibition of Autotaxin Is Efficacious in Mouse Models of Liver Fibrosis. J. Pharmacol. Exp. Ther. 2017, 360, 1–13.
- TaiwanJ Pharmaceuticals. Available online: https://www.taiwanj.com/pages/page_index_en (accessed on 10 December 2021).
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; M Rodrigues, R. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2019, 9, 37.
- Athyros, V.G.; Mikhailidis, D.P.; Didangelos, T.P.; Giouleme, O.I.; Liberopoulos, E.N.; Karagiannis, A.; Kakafika, A.I.; Tzi-omalos, K.; Burroughs, A.K.; Elisaf, M.S. Effect of multifactorial treatment on non-alcoholic fatty liver disease in metabolic syndrome: a randomised study. Curr Med Res Opin 2006, 22, 873–83.
- Fernández-Miranda, C.; Pérez-Carreras, M.; Colina, F.; López-Alonso, G.; Vargas, C.; Solís-Herruzo, J.A. A pilot trial of feno-fibrate for the treatment of non-alcoholic fatty liver disease. Dig Liver Dis 2008, 40, 200–5.
- Basaranoglu, M.; Acbay, O.; Sonsuz, A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic stea-tohepatitis. J Hepatol 1999, 31, 384.
- Laurin, J.; Lindor, K.D.; Crippin, J.S.; Gossard, A.; Gores, G.J.; Ludwig, J.; Rakela, J.; McGill, D.B. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: a pilot study. Hepatology 1996, 23, 1464–7.
- Hatanaka, T.; Kosone, T.; Saito, N.; Takakusagi, S.; Tojima, H.; Naganuma, A.; Takagi, H.; Uraoka, T.; Kakizaki, S. Effect of 48-week pemafibrate on non-alcoholic fatty liver disease with hypertriglyceridemia, as evaluated by the FibroScan-aspartate aminotransferase score. JGH Open 2021, 5, 1183–1189.
- Sasaki, Y.; Shimada, T.; Iizuka, S.; Suzuki, W.; Makihara, H.; Teraoka, R.; Tsuneyama, K.; Hokao, R.; Aburada, M. Effects of bezafibrate in nonalcoholic steatohepatitis model mice with monosodium glutamate-induced metabolic syndrome. Eur J Pharmacol 2011, 662, 1–8.
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357.
- Lee, Y.H.; Kim, J.H.; Kim, S.R.; Jin, H.Y.; Rhee, E.J.; Cho, Y.M.; Lee, B.W. Lobeglitazone, a Novel Thiazolidinedione, Improves Non-Alcoholic Fatty Liver Disease in Type 2 Diabetes: Its Efficacy and Predictive Factors Related to Responsiveness. J Korean Med Sci 2017, 32, 60–69.
- Nakagami, H.; Shimamura, M.; Miyake, T.; Shimosato, T.; Minobe, N.; Moritani, T.; Kiomy Osako, M.; Nakagami, F.; Koriyama, H.; Kyutoku, M.; Shimizu, H.; Katsuya, T.; Morishita, R. Nifedipine prevents hepatic fibrosis in a non-alcoholic steatohepatitis model induced by an L-methionine-and choline-deficient diet. Mol Med Rep 2012, 5, 37–40.
- Zarei, M.; Barroso, E.; Palomer, X.; Dai, J.; Rada, P.; Quesada-López, T.; Escolà-Gil, J.C.; Cedó, L.; Zali, M.R.; Molaei, M.; Dabiri, R.; Vázquez, S.; Pujol, E.; Valverde, Á.M.; Villarroya, F.; Liu, Y.; Wahli, W.; Vázquez-Carrera, M. Hepatic regulation of VLDL receptor by PPARβ/δ and FGF21 modulates non-alcoholic fatty liver disease. Mol Metab 2018, 8, 117–131.
- CymaBay Therapeutics. CymaBay Therapeutics Reports Topline 12-Week Data from an Ongoing Phase 2b Study of Seladelpar in Patients with Nonalcoholic Steatohepatitis, 2019.
- Goyal, O.; Nohria, S.; Goyal, P.; Kaur, J.; Sharma, S.; Sood, A.; Chhina, R.S. Saroglitazar in patients with non-alcoholic fatty liver disease and diabetic dyslipidemia: a prospective, observational, real world study. Sci Rep 2020, 10, 21117.
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.; Kowdley, K.V.; Lai, M.; Schiff, E.; Parmar, D.; Patel, P.; Chalasani, N. Saroglitazar, a PPAR-α/γ Agonist, for Treatment of NAFLD: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology 2021, 74, 1809–1824.
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; Lanthier, N.; Alkhouri, N.; Moreno, C.; Schattenberg, J.M.; Stefanova-Petrova, D.; Vonghia, L.; Rouzier, R.; Guillaume, M.; Hodge, A.; Romero-Gómez, M.; Huot-Marchand, P.; Baudin, M.; Richard, M.P.; Abitbol, J.L.; Broqua, P.; Junien, J.L.; Abdelmalek, M.F.; NATIVE Study Group. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N Engl J Med 2021, 385, 1547–1558.
- Harrison, S.A.; Alkhouri, N.; Davison, B.A.; Sanyal, A.; Edwards, C.; Colca, J.R.; Lee, B.H.; Loomba, R.; Cusi, K.; Kolterman, O.; Cotter, G.; Dittrich, H.C. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study. J Hepatol. 2020 Apr;72(4):613-626.
- Fukunaga, T.; Zou, W.; Rohatgi, N.; Colca, J.R.; Teitelbaum, S.L. An insulin-sensitizing thiazolidinedione, which minimally activates PPARγ, does not cause bone loss. J Bone Miner Res 2015, 30, 481–8.
- Jacques, V.; Bolze, S.; Hallakou-Bozec, S.; Czarnik, A.W.; Divakaruni, A.S.; Fouqueray, P.; Murphy, A.N.; Van der Ploeg, L.H.T.; DeWitt, S. Deuterium-Stabilized (R)-Pioglitazone (PXL065) Is Responsible for Pioglitazone Efficacy in NASH yet Ex-hibits Little to No PPARγ Activity. Hepatol Commun 2021, 5, 1412–1425.
- El, K.; Gray, S.M.; Capozzi, M.E.; Knuth, E.R.; Jin, E.; Svendsen, B.; Clifford, A.; Brown, J.L.; Encisco, S.E.; Chazotte, B.M.; Sloop, K.W.; Nunez, D.J.; Merrins, M.J.; D'Alessio, D.A.; Campbell, J.E. GIP mediates the incretin effect and glucose tolerance by dual actions on α cells and β cells. Sci Adv 2021, 7, eabf1948.
- Wang, X.C.; Gusdon, A.M.; Liu, H.; Qu, S. Effects of glucagon-like peptide-1 receptor agonists on non-alcoholic fatty liver disease and inflammation. World J Gastroenterol 2014, 20, 14821–30.
- Samson, S.L.; Sathyanarayana, P.; Jogi, M.; Gonzalez, E.V.; Gutierrez, A.; Krishnamurthy, R.; Muthupillai, R.; Chan, L.; Bajaj, M. Exenatide decreases hepatic fibroblast growth factor 21 resistance in non-alcoholic fatty liver disease in a mouse model of obesity and in a randomised controlled trial. Diabetologia 2011, 54, 3093–100.
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.S.; Harrison, S.A.; NN9931-4296 Investigators. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N Engl J Med 2021, 384, 1113–1124.
- Liava, C.; Sinakos, E. Semaglutide for nonalcoholic steatohepatitis: closer to a solution? Hepatobiliary Surg Nutr 2021, 10, 541–544.
- Dutour, A.; Abdesselam, I.; Ancel, P.; Kober, F.; Mrad, G.; Darmon, P.; Ronsin, O.; Pradel, V.; Lesavre, N.; Martin, J.C.; Jacquier, A.; Lefur, Y.; Bernard, M.; Gaborit, B. Exenatide decreases liver fat content and epicardial adipose tissue in patients with obesity and type 2 diabetes: a prospective randomized clinical trial using magnetic resonance imaging and spectroscopy. Diabetes Obes Metab 2016, 18, 882–91.
- Sofogianni, A.; Filippidis, A.; Chrysavgis, L.; Tziomalos, K.; Cholongitas, E. Glucagon-like peptide-1 receptor agonists in non-alcoholic fatty liver disease: An update. World J Hepatol 2020, 12, 493–505.
- Cusi, K.; Sattar, N.; García-Pérez, L.E.; Pavo, I.; Yu, M.; Robertson, K.E.; Karanikas, C.A.; Haupt, A. Dulaglutide decreases plasma aminotransferases in people with Type 2 diabetes in a pattern consistent with liver fat reduction: a post hoc analysis of the AWARD programme. Diabet Med 2018, 35, 1434–1439.
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Choudhary, N.S.; Singh, M.K.; Wasir, J.S.; Kaur, P.; Gill, H.K.; Bano, T.; Farooqui, K.J.; Mithal, A. Effect of dulaglutide on liver fat in patients with type 2 diabetes and NAFLD: randomised controlled trial (D-LIFT trial). Diabetologia 2020, 63, 2434–2445.
- Koutsovasilis, A.; Sotiropoulos, A.; Pappa, M.; Apostolou, O.; Binikos, I.; Kordinas, V.; Papadaki, D.; Tamvakos, I.; Bousboulas, S. Effectiveness of lixisenatide in nonalcoholic fatty liver disease in patients with type 2 diabetes after an acute coronary syn-drome compared to sitagliptin and pioglitazone. In Proceedings of the EASD Virtual Meeting 2016, Munich, Germany, 14 Sep 2016.
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; LEAN trial team; Abouda, G.; Aldersley, M.A.; Stocken, D.; Gough, S.C.; Tomlinson, J.W.; Brown, R.M.; Hübscher, S.G.; Newsome, P.N. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690.
- Ghosal, S.; Datta, D.; Sinha, B. A meta-analysis of the effects of glucagon-like-peptide 1 receptor agonist (GLP1-RA) in non-alcoholic fatty liver disease (NAFLD) with type 2 diabetes (T2D). Sci Rep 2021, 11, 22063.
- Thondam, S.K.; Cuthbertson, D.J.; Wilding, J.P.H. The influence of Glucose-dependent Insulinotropic Polypeptide (GIP) on human adipose tissue and fat metabolism: Implications for obesity, type 2 diabetes and Non-Alcoholic Fatty Liver Disease (NAFLD). Peptides 2020, 125, 170208.
- Hartman, M.L.; Sanyal, A.J.; Loomba, R.; Wilson, J.M.; Nikooienejad, A.; Bray, R.; Karanikas, C.A.; Duffin, K.L.; Robins, D.A.; Haupt, A. Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes. Diabetes Care 2020, 43, 1352–1355.
- Gastaldelli, A.; Cusi, K.; Landó, F.; Bray, R.; Brouwers, B.; Rodríguez, Á. Effect of tirzepatide versus insulin degludec on liver fat content and abdominal adipose tissue in patients with type 2 diabetes (SURPASS-3 MRI). In Proceedings of the EASD Virtual Meeting 2021, Virtual, 30 Sep 2021.
- Pocai, A. Action and therapeutic potential of oxyntomodulin. Mol Metab 2013, 3, 241–51.
- Boland, M.L.; Laker, R.C.; Mather, K.; Nawrocki, A.; Oldham, S.; Boland, B.B.; Lewis, H.; Conway, J.; Naylor, J.; Guionaud, S.; Feigh, M.; Veidal, S.S.; Lantier, L.; McGuinness, O.P.; Grimsby, J.; Rondinone, CM..; Jermutus, L.; Larsen, M.R.; Trevaskis, J.L.; Rhodes, C.J. Resolution of NASH and hepatic fibrosis by the GLP-1R/GcgR dual-agonist Cotadutide via modulating mito-chondrial function and lipogenesis. Nat Metab 2020, 2, 413–431.
- Nahra, R.; Wang, T.; Gadde, K.M.; Oscarsson, J.; Stumvoll, M.; Jermutus, L.; Hirshberg, B.; Ambery, P. Effects of Cotadutide on Metabolic and Hepatic Parameters in Adults With Overweight or Obesity and Type 2 Diabetes: A 54-Week Randomized Phase 2b Study. Diabetes Care 2021, 44, 1433–1442.
- Jung, S.Y.; Park, Y.J.; Lee, J.S.; Kim, E.J.; Lee, Y.M.; Kim, Y.H.; Trautmann, M.; Kwon, S.C. Potent Cholesterol Lowering Effect by HM12525A, A Novel Long-acting GLP-1/Glucagon Dual Receptor Agonist. In Proceedings of the American Diabetes As-sociation’s (ADA) 76th Scientific Sessions, New Orleans (LA), USA, 10-14 Jun 2016.
- Pfizer Inc. Available online: https://www.pfizer.com/ (accessed on 10 Dec 2021).
- Hanmi Pharmaceutical Co.; Ltd. Available online: http://www.hanmipharm.com/ehanmi/handler/Home-Start (accessed on 10 Dec 2021).
- Kim, J.K.; Lee, J.S.; Park, E.; Lee, J.; Bae, S.; Kim, D.; Choi, I.C. HM15211, a Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist, Exhibits Anti-inflammatory and Fibrotic Effects in AMLN/TAA–Induced Liver Inflammation and Fibrosis Mice. Diabetes 2020, 69, 1804-P.
- Janssen, P.; Rotondo, A.; Mulé, F.; Tack, J. Review article: a comparison of glucagon-like peptides 1 and 2. Aliment Pharmacol Ther 2013, 37, 18–36.
- Hu, Y.-X. Therapeutic effect of teduglutide on non-alcoholic fatty liver disease in rats. World Chin J Dig 2016, 24, 1009.
- Alam, S.; Ghosh, J.; Mustafa, G.; Kamal, M.; Ahmad, N. Effect of sitagliptin on hepatic histological activity and fibrosis of nonalcoholic steatohepatitis patients: a 1-year randomized control trial. Hepat Med 2018, 10, 23–31.
- Aktaş, A.; Ozan, Z.T. The efficacy and safety of vildagliptin treatment for nonalcoholic fatty liver disease in type 2 diabetes mellitus. Cumhur Medical J 2020, 42, 491–499.
- Li, J.J.; Zhang, P.; Fan, B.; Guo, X.L.; Zheng, Z.S. The efficacy of saxagliptin in T2DM patients with non-alcoholic fatty liver disease: preliminary data. Rev Assoc Med Bras (1992) 2019, 65, 33–37.
- Hattori, S.; Nomoto, K.; Suzuki, T.; Hayashi, S. Beneficial effect of omarigliptin on diabetic patients with non-alcoholic fatty liver disease/non-alcoholic steatohepatitis. Diabetol Metab Syndr 2021, 13, 28.
- Gupta, V.K. Teneligliptin Significantly Reduces Liver Fat Content (LFC) and Delays Progression of NASH in Type 2 Diabetes Mellitus Patients. Diabetes 2019, 68, 1029-P.
- Mashitani, T.; Noguchi, R.; Okura, Y.; Namisaki, T.; Mitoro, A.; Ishii, H.; Nakatani, T.; Kikuchi, E.; Moriyasu, H.; Matsumoto, M.; Sato, S.; An, T.; Morita, H.; Aizawa, S.; Tokuoka, Y.; Ishikawa, M.; Matsumura, Y.; Ohira, H.; Kogure, A.; Noguchi, K.; Yoshiji, H. Efficacy of alogliptin in preventing non-alcoholic fatty liver disease progression in patients with type 2 diabetes. Biomed Rep 2016, 4, 183–187.
- Dos Santos, L.R.; Duarte, M.L.; Peccin, M.S.; Gagliardi, A.R.T, Melnik, T. Dipeptidyl Peptidase IV Inhibitors for Nonalcoholic Fatty Liver Disease – Systematic Review and Metanalysis. Curr Diabetes Rev 2021, 17, e101120187811.
- Wang, Z.; Park, H.; Bae, E.J. Efficacy of evogliptin and cenicriviroc against nonalcoholic steatohepatitis in mice: a comparative study. Korean J Physiol Pharmacol 2019, 23, 459–466.
- Sakai, Y.; Nagashimada, M.; Nagata, N.; Ni, Y.; Kaneko, S.; Ota, T. Dipeptidyl peptidase-4 inhibitor anagliptin alleviates lipotoxicity-induced hepatic insulin resistance and steatohepatitis in mice. In Proceedings of the EASD Virtual Meeting 2015, Stockholm, Sweden, 15 Sep 2015.
- Kawakubo, M.; Tanaka, M.; Ochi, K.; Watanabe.; A.; Saka-Tanaka.; M.; Kanamori, Y.; Yoshioka, N.; Yamashita, S.; Goto, M.; Itoh, M.; Shirakawa, I.; Kanai, S.; Suzuki, H.; Sawada, M.; Ito, A.; Ishigami, M.; Fujishiro, M.; Arima, H.; Ogawa, Y.; Suganami, T. Dipeptidyl peptidase-4 inhibition prevents nonalcoholic steatohepatitis-associated liver fibrosis and tumor development in mice independently of its anti-diabetic effects. Sci Rep 2020, 10, 983.
- Wang, G.; Wu, B.; Zhang, L.; Jin, X.; Wang, K.; Xu, W.; Zhang, B.; Wang, H. The protective effects of trelagliptin on high-fat diet-induced nonalcoholic fatty liver disease in mice. J Biochem Mol Toxicol 2021, 35, e22696.
- Choi, S.H, Leem, J.; Park, S.; Lee, C.K.; Park, K.G.; Lee, I.K. Gemigliptin ameliorates Western-diet-induced metabolic syndrome in mice. Can J Physiol Pharmacol 2017, 95, 129–139.
- Klein, T.; Fujii, M.; Sandel, J.; Shibazaki, Y.; Wakamatsu, K.; Mark, M.; Yoneyama, H. Linagliptin alleviates hepatic steatosis and inflammation in a mouse model of non-alcoholic steatohepatitis. Med Mol Morphol 2014, 47, 137–49.
- Saisho, Y. SGLT2 Inhibitors: the Star in the Treatment of Type 2 Diabetes? Diseases 2020, 8, 14.
- Wu, P.; Wen, W.; Li, J.; Xu, J.; Zhao, M.; Chen, H.; Sun, J. Systematic Review and Meta-Analysis of Randomized Controlled Trials on the Effect of SGLT2 Inhibitor on Blood Leptin and Adiponectin Level in Patients with Type 2 Diabetes. Horm Metab Res 2019, 51, 487–494.
- Sumida, Y.; Yoneda, M.; Tokushige, K.; Kawanaka, M.; Fujii, H.; Yoneda, M.; Imajo, K.; Takahashi, H.; Ono, M.; Nozaki, Y.; Hyogo, H.; Koseki, M.; Yoshida, Y.; Kawaguchi, T.; Kamada, Y.; Eguchi, Y.; Okanoue, T.; Nakajima, A.; Japan Study Group of NAFLD (JSG-NAFLD). Hepatoprotective Effect of SGLT2 Inhibitor on Nonalcoholic Fatty Liver Disease. Diab Res Open Access 2020, 2, 17–25.
- Ribeiro Dos Santos, L, Baer Filho, R. Treatment of nonalcoholic fatty liver disease with dapagliflozin in non-diabetic patients. Metabol Open 2020, 5, 100028.
- Taheri, H.; Malek, M.; Ismail-Beigi, F.; Zamani, F.; Sohrabi, M.; Reza Babaei, M.; Khamseh, M.E. Effect of Empagliflozin on Liver Steatosis and Fibrosis in Patients With Non-Alcoholic Fatty Liver Disease Without Diabetes: A Randomized, Double-Blind, Placebo-Controlled Trial. Adv Ther 2020, 37, 4697–4708.
- Itani, T.; Ishihara, T. Efficacy of canagliflozin against nonalcoholic fatty liver disease: a prospective cohort study. Obes Sci Pract 2018, 4, 477–482.
- Prikhodko, V.A.; Sysoev, Yu.I.; Poveryaeva, M.A.; Bunyat, A.V.; Karev, V.E.; Ivkin, D.Yu.; Sukhanov, D.S.; Shustov, E.B.; Okovityi, S.V. Effects of Empagliflozin and L-Ornithine L-Aspartate on Behavior, Cognitive Functions, and Physical Perfor-mance in Mice with Experimentally Induced Steatohepatitis. Bull RSMU 2020, 3, 49–57.
- Takahashi, H.; Kessoku, T.; Kawanaka, M.; Nonaka, M.; Hyogo, H.; Fujii, H.; Nakajima, T.; Imajo, K.; Tanaka, K.; Kubotsu, Y.; Isoda, H.; Oeda, S.; Kurai, O.; Yoneda, M.; Ono, M.; Kitajima, Y.; Tajiri, R.; Takamori, A.; Kawaguchi, A.; Aishima, S.; Kage, M.; Nakajima, A.; Eguchi, Y.; Anzai, K. Ipragliflozin Improves the Hepatic Outcomes of Patients With Diabetes with NAFLD. Hepatol Commun 2021, online ahead of print.
- Wilkison, B.; Cheatham, B.; Walker, S. Remogliflozin Etabonate Reduces FIB-4 and NAFLD Fibrosis Scores in Type 2 Diabetic Subjects. Hepatology 2016, 64, 548A.
- Gallo, S.; Calle, R.A.; Terra, S.G.; Pong, A.; Tarasenko, L.; Raji, A. Effects of Ertugliflozin on Liver Enzymes in Patients with Type 2 Diabetes: A Post-Hoc Pooled Analysis of Phase 3 Trials. Diabetes Ther 2020, 11, 1849–1860.
- Shibuya, T.; Fushimi, N.; Kawai, M.; Yoshida, Y.; Hachiya, H.; Ito, S.; Kawai, H.; Ohashi, N.; Mori, A. Luseogliflozin improves liver fat deposition compared to metformin in type 2 diabetes patients with non-alcoholic fatty liver disease: A prospective randomized controlled pilot study. Diabetes Obes Metab 2018, 20, 438–442.
- Sumida, Y.; Murotani, K.; Saito, M.; Tamasawa, A.; Osonoi, Y.; Yoneda, M.; Osonoi, T. Effect of luseogliflozin on hepatic fat content in type 2 diabetes patients with non-alcoholic fatty liver disease: A prospective, single-arm trial (LEAD trial). Hepatol Res 2019, 49, 64–71.
- Yoneda, M.; Honda, Y.; Ogawa, Y.; Kessoku, T.; Kobayashi, T.; Imajo, K.; Ozaki, A.; Nogami, A.; Taguri, M.; Yamanaka, T.; Kirikoshi, H.; Iwasaki, T.; Kurihashi, T.; Saito, S.; Nakajima, A. Comparing the effects of tofogliflozin and pioglitazone in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus (ToPiND study): a randomized prospective open-label controlled trial. BMJ Open Diabetes Res Care 2021, 9, e001990.
- He, Y.L.; Haynes, W.; Meyers, C.D.; Amer, A.; Zhang, Y.; Mahling, P.; Mendonza, A.E.; Ma, S.; Chutkow, W.; Bachman, E. The effects of licogliflozin, a dual SGLT1/2 inhibitor, on body weight in obese patients with or without diabetes. Diabetes Obes Metab 2019, 21, 1311–1321.
- Harrison, S.A.; Manghi, F.P.; Smith, W.B.; Alpenidze, D.; Aizenberg, D.; Burggraaf, K.; Chen, C.-Y.; Zuckerman, E.; Ravussin, E.; Charatcharoenwitthaya, P.; Cheng II, P.-N.; Katchman, H.; Klein, S.; Ben-Ari, Z.; Mendonza, A.; Zhang, Y.; Martic, M.; Ma, S.; Kao, S.; Tanner, S.; Pachori, A.; He, Y.-L.; Ukomadu, C.; Sicard, E. LIK066 (Licogliflozin), an SGLT1/2 Inhibitor, Robustly Decreases ALT and Improves Markers of Hepatic and Metabolic Health in Patients with Non-Alcoholic Fatty Liver Disease: Interim Analysis of a 12-Week, Randomized, Placebo-Controlled, Phase 2a Study. In Proceedings of the Liver Meeting 2019, Boston (MA), USA, 8-12 Nov 2019.
- Honda, Y.; Ozaki, A.; Iwaki, M.; Kobayashi, T.; Nogami, A.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Yoneda, M.; Saito, S.; Nagashima, Y.; Nakajima, A. Protective effect of SGL5213, a potent intestinal sodium-glucose cotransporter 1 inhibitor, in nonalcoholic fatty liver disease in mice. J Pharmacol Sci 2021, 147, 176-183.
- Iogna Prat, L.; Tsochatzis, E.A. The effect of antidiabetic medications on non-alcoholic fatty liver disease (NAFLD). Hormones (Athens) 2018, 17, 219–229.
- Hajiaghamohammadi, A.A.; Miroliaee, A.; Samimi, R.; Alborzi, F.; Ziaee, A. A Comparison of Ezetimibe and Acarbose in Decreasing Liver Transaminase in Nonalcoholic Fatty Liver Disease: A Randomized Clinical Trial. Govaresh 2013, 18, 186–90.
- Komatsu, M.; Tanaka, N.; Kimura, T.; Fujimori, N.; Sano, K.; Horiuchi, A.; Sugiura, A.; Yamazaki, T.; Shibata, S.; Joshita, S.; Umemura, T.; Matsumoto, A.; Tanaka, E. Miglitol attenuates non-alcoholic steatohepatitis in diabetic patients. Hepatol Res 2018, 48, 1092–1098.
- Kim, J.W.; Lee, Y.J.; You, Y.H.; Moon, M.K.; Yoon, K.H.; Ahn, Y.B.; Ko, S.H. Effect of sodium-glucose cotransporter 2 inhibitor, empagliflozin, and α-glucosidase inhibitor, voglibose, on hepatic steatosis in an animal model of type 2 diabetes. J Cell Biochem 2018, online ahead of print.
- Meroni, M.; Longo, M.; Dongiovanni, P. The Role of Probiotics in Nonalcoholic Fatty Liver Disease: A New Insight into Therapeutic Strategies. Nutrients 2019, 11, 2642.
- Malaguarnera, M.; Vacante, M.; Antic, T.; Giordano, M.; Chisari, G.; Acquaviva, R.; Mastrojeni, S.; Malaguarnera, G.; Mis-tretta, A.; Li Volti, G.; Galvano, F. Bifidobacterium longum with fructo-oligosaccharides in patients with non alcoholic stea-tohepatitis. Dig Dis Sci 2012, 57, 545-53.
- Vajro, P.; Mandato, C.; Licenziati, M.R.; Franzese, A.; Vitale, D.F.; Lenta, S.; Caropreso, M.; Vallone, G.; Meli, R. Effects of Lactobacillus rhamnosus strain GG in pediatric obesity-related liver disease. J Pediatr Gastroenterol Nutr 2011, 52, 740–3.
- Nabavi, S.; Rafraf, M.; Somi, M.H.; Homayouni-Rad, A.; Asghari-Jafarabadi, M. Effects of probiotic yogurt consumption on metabolic factors in individuals with nonalcoholic fatty liver disease. J Dairy Sci 2014, 97, 7386–93.
- Román, E.; Nieto, J.C.; Gely, C.; Vidal, S.; Pozuelo, M.; Poca, M.; Juárez, C.; Guarner, C.; Manichanh, C.; Soriano, G. Effect of a Multistrain Probiotic on Cognitive Function and Risk of Falls in Patients With Cirrhosis: A Randomized Trial. Hepatol Commun 2019, 3, 632–645.
- Ahn, S.B.; Jun, D.W.; Kang, B.K.; Lim, J.H.; Lim, S.; Chung, M.J. Randomized, Double-blind, Placebo-controlled Study of a Multispecies Probiotic Mixture in Nonalcoholic Fatty Liver Disease. Sci Rep 2019, 9, 5688.
- Kobyliak, N.; Abenavoli, L.; Mykhalchyshyn, G.; Kononenko, L.; Boccuto, L.; Kyriienko, D.; Dynnyk, O. A Multi-strain Pro-biotic Reduces the Fatty Liver Index, Cytokines and Aminotransferase levels in NAFLD Patients: Evidence from a Randomized Clinical Trial. J Gastrointestin Liver Dis 2018, 27, 41–49.
- Ma, Y.Y.; Li, L.; Yu, C.H.; Shen, Z.; Chen, L.H.; Li, Y.M. Effects of probiotics on nonalcoholic fatty liver disease: a meta-analysis. World J Gastroenterol 2013, 19, 6911–8.
- Gao, X.; Zhu, Y.; Wen, Y.; Liu, G.; Wan, C. Efficacy of probiotics in non-alcoholic fatty liver disease in adult and children: A meta-analysis of randomized controlled trials. Hepatol Res 2016, 46, 1226–1233.
- Liu, Y.T.; Li, Y.Q.; Wang, Y.Z. [Protective effect of Saccharomyces boulardii against intestinal mucosal barrier injury in rats with nonalcoholic fatty liver disease]. Zhonghua Gan Zang Bing Za Zhi 2016, 24, 921-926. Chinese.
- Yamauchi, R.; Takedatsu, H.; Yokoyama, K.; Yamauchi, E.; Kawashima, M.; Tsuchiya, N.; Takata, K.; Tanaka, T.; Morihara, D.; Takeyama, Y.; Shakado, S.; Sakisaka, S.; Hirai, F. Synergistic effect of clostridium butyricum miyairi on rifaximin in mice model of non-alcoholic steatohepatitis by methionine choline-deficient diet. J Hepatol 2020, 73, S401–S652.
- Ezquer, M.; Ezquer, F.; Ricca, M.; Allers, C.; Conget, P. Intravenous administration of multipotent stromal cells prevents the onset of non-alcoholic steatohepatitis in obese mice with metabolic syndrome. J Hepatol 2011, 55, 1112–20.
- Hsu, M.J.; Karkossa, I.; Schäfer, I.; Christ, M.; Kühne, H.; Schubert, K.; Rolle-Kampczyk, U.E.; Kalkhof, S.; Nickel, S.; Seibel, P.; von Bergen, M.; Christ, B. Mitochondrial Transfer by Human Mesenchymal Stromal Cells Ameliorates Hepatocyte Lipid Load in a Mouse Model of NASH. Biomedicines 2020, 8, 350.
- Wang, H.; Wang, D.; Yang, L.; Wang, Y.; Jia, J.; Na, D.; Chen, H.; Luo, Y.; Liu, C. Compact bone-derived mesenchymal stem cells attenuate nonalcoholic steatohepatitis in a mouse model by modulation of CD4 cells differentiation. Int Immunopharmacol. 2017, 42, 67–73.
- Chien, Y.; Huang, C.S.; Lin, H.C.; Lu, K.H.; Tsai, P.H.; Lai, Y.H.; Chen, K.H.; Lee, S.D.; Huang, Y.H.; Wang, C.Y. Improvement of non-alcoholic steatohepatitis by hepatocyte-like cells generated from iPSCs with Oct4/Sox2/Klf4/Parp1. Oncotarget 2018, 9, 18594–18606.
- Overi, D.; Carpino, G.; Franchitto, A.; Onori, P.; Gaudio, E. Hepatocyte Injury and Hepatic Stem Cell Niche in the Progression of Non-Alcoholic Steatohepatitis. Cells 2020, 9, 590.
- Nevens, F.; Gustot, T.; Laterre, P.F.; Lasser, L.L.; Haralampiev, L.E.; Vargas, V.; Lyubomirova, D.; Albillos, A.; Najimi, M.; Michel, S.; Stoykov, I.; Gordillo, N.; Vainilovich, Y.; Barthel, V.; Clerget-Chossat, N.; Sokal, E.M. A phase II study of human allogeneic liver-derived progenitor cell therapy for acute-on-chronic liver failure and acute decompensation. JHEP Rep 2021, 3, 100291.
- Iqbal, U.; Perumpail, B.J.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. Judicious Use of Lipid Lowering Agents in the Management of NAFLD. Diseases 2018, 6, 87.
- Oniciu, D.C.; Hashiguchi, T.; Shibazaki, Y.; Bisgaier, C.L. Gemcabene downregulates inflammatory, lipid-altering and cell-signaling genes in the STAM™ model of NASH. PLoS One 2018, 13, e0194568.
- Sanjay, K.V.; Vishwakarma, S.; Zope.; B.R.; Mane, V.S.; Mohire, S.; Dhakshinamoorthy, S. ATP citrate lyase inhibitor Bem-pedoic Acid alleviate long term HFD induced NASH through improvement in glycemic control, reduction of hepatic triglyc-erides & total cholesterol, modulation of inflammatory & fibrotic genes and improvement in NAS score. CRPHAR 2021, 2, 100051.
- Stanley, T.L.; Feldpausch, M.N.; Oh, J.; Branch, K.L.; Lee, H.; Torriani, M.; Grinspoon, S.K. Effect of tesamorelin on visceral fat and liver fat in HIV-infected patients with abdominal fat accumulation: a randomized clinical trial. JAMA 2014, 312, 380–9.
- Theratechnologies Inc. Available online: https://www.theratech.com/ (accessed on 10 Dec 2021).
- Wei, X.; Wang, C.; Hao, S.; Song, H.; Yang, L. The Therapeutic Effect of Berberine in the Treatment of Nonalcoholic Fatty Liver Disease: A Meta-Analysis. Evid Based Complement Alternat Med 2016, 2016, 3593951.
- Yin, J.; Ye, J.; Jia, W. Effects and mechanisms of berberine in diabetes treatment. Acta Pharm Sin B 2012, 2, 327–334.
- Simental-Mendía, M.; Sánchez-García, A.; Simental-Mendía, L.E. Effect of ursodeoxycholic acid on liver markers: A systematic review and meta-analysis of randomized placebo-controlled clinical trials. Br J Clin Pharmacol 2020, 86, 1476–1488.
- Zhang, W.; Tang, Y.; Huang, J.; Hu, H. Efficacy of ursodeoxycholic acid in nonalcoholic fatty liver disease: An updated me-ta-analysis of randomized controlled trials. Asia Pac J Clin Nutr 2020, 29, 696–705.
- Harrison, S.A.; Gunn, N.; Neff, G.W.; Kohli, A.; Liu, L.; Flyer, A.; Goldkind, L.; Di Bisceglie, A.M. A phase 2, proof of concept, randomised controlled trial of berberine ursodeoxycholate in patients with presumed non-alcoholic steatohepatitis and type 2 diabetes. Nat Commun 2021, 12, 5503.
- Kowdley, K.V.; Butler, P.; Cubberley, S.; Hand, A.L.; Jenders, R.A.; Kroon, J.; Leibowitz, M.; Moore, A.C.; Guyer, B. Miricorilant, a Selective GR Modulator, Induced a Rapid and Significant Reduction in Liver Fat Content in a Randomized, Placebo-Controlled Phase 2a Study in Patients with Non-Alcoholic Steatohepatitis. In Proceedings of the Liver Meeting 2021, Virtual, 12-15 Nov 2021.
- Li, F.; Jiang, M.; Ma, M.; Chen, X.; Zhang, Y.; Zhang, Y.; Yu, Y.; Cui, Y.; Chen, J.; Zhao, H.; Sun, Z.; Dong, D. Anthelmintics nitazoxanide protects against experimental hyperlipidemia and hepatic steatosis in hamsters and mice. Acta Pharm Sin B 2021, in press, corrected proof.
- Walczak, R.; Carole, B.; Benoit, N.; Descamps, E.; Nathalie, D.; Meghien, S.; Hum, D.; Staels, B.; Friedman, S.; Loomba, R.; Harrison, S.A. Elafibranor and nitazoxanide synergize to reduce fibrosis in a NASH model. J Hepatol 2018, 68, S105-S364.
- Glal, K.A.M.; Abd-Elsalam, S.M.; Mostafa, T.M. Nitazoxanide versus rifaximin in preventing the recurrence of hepatic en-cephalopathy: A randomized double-blind controlled trial. J Hepatobiliary Pancreat Sci 2021, 28, 812–824.
- Flores-Contreras, L.; Sandoval-Rodríguez, A.S.; Mena-Enriquez, M.G.; Lucano-Landeros, S.; Arellano-Olivera, I.; Alva-rez-Álvarez, A.; Sanchez-Parada, M.G.; Armendáriz-Borunda J. Treatment with pirfenidone for two years decreases fibrosis, cytokine levels and enhances CB2 gene expression in patients with chronic hepatitis C. BMC Gastroenterol 2014, 14, 131.
- Cui, Y.; Zhang, M.; Leng, C.; Blokzijl, T.; Jansen, B.H.; Dijkstra, G.; Faber, K.N. Pirfenidone Inhibits Cell Proliferation and Collagen I Production of Primary Human Intestinal Fibroblasts. Cells 2020, 9, 775.
- Poo, J.L.; Torre, A.; Aguilar-Ramírez, J.R.; Cruz, M.; Mejía-Cuán, L.; Cerda, E.; Velázquez, A.; Patiño, A.; Ramírez-Castillo, C.; Cisneros, L.; Bosques-Padilla, F.; Hernández, L.; Gasca, F.; Flores-Murrieta, F.; Treviño, S.; Tapia, G.; Armendariz-Borunda, J.; Muñoz-Espinosa, L.E. Benefits of prolonged-release pirfenidone plus standard of care treatment in patients with advanced liver fibrosis: PROMETEO study. Hepatol Int 2020, 14, 817–827.
- Matsuda, K.; Iwaki, Y. MN-001 (tipelukast), a novel, orally bioavailable drug, reduces fibrosis and inflammation and down-regulates TIMP-1, collagen Type 1 and LOXL2 mRNA overexpression in an advanced NASH (non-alcoholic steato-hepatitis) model. In Proceedings of the Liver Meeting 2014, Boston (MA), USA, 07-11 Nov 2014.
- Yang, M.; Zhang, C.Y. G protein-coupled receptors as potential targets for nonalcoholic fatty liver disease treatment. World J Gastroenterol 2021, 27, 677–691.
- Widjaja, A.A.; Singh, B.K.; Adami, E.; Viswanathan, S.; Dong, J.; D'Agostino, G.A.; Ng, B.; Lim.; W.W.; Tan, J.; Paleja, B.S.; Tripathi, M.; Lim, S.Y.; Shekeran, S.G.; Chothani, S.P.; Rabes, A.; Sombetzki, M.; Bruinstroop, E.; Min, L.P.; Sinha, R.A.; Albani, S.; Yen, P.M.; Schafer, S.; Cook, S.A. Inhibiting Interleukin 11 Signaling Reduces Hepatocyte Death and Liver Fibrosis, In-flammation, and Steatosis in Mouse Models of Nonalcoholic Steatohepatitis. Gastroenterology 2019, 157, 777–792.e14.
- Zai, W.; Chen, W.; Liu, H.; Ju, D. Therapeutic Opportunities of IL-22 in Non-Alcoholic Fatty Liver Disease: From Molecular Mechanisms to Clinical Applications. Biomedicines 2021, 9, 1912.
- Stemmer, S.M.; Manojlovic, N.S.; Marinca, M.V.; Petrov, P.; Cherciu, N.; Ganea, D.; Ciuleanu, T.E.; Pusca, I.A.; Beg, M.S.; Purcell, W.T.; Croitoru, A.E.; Ilieva, R.N.; Natošević, S.; Nita, A.L.; Kalev, D.N.; Harpaz, Z.; Farbstein, M.; Silverman, M.H.; Bristol, D.; Itzhak, I.; Fishman, P. Namodenoson in Advanced Hepatocellular Carcinoma and Child-Pugh B Cirrhosis: Ran-domized Placebo-Controlled Clinical Trial. Cancers (Basel) 2021, 13, 187.
- Ferramosca, A.; Di Giacomo, M.; Zara, V. Antioxidant dietary approach in treatment of fatty liver: New insights and updates. World J Gastroenterol 2017, 23, 4146–4157.
- Ionis Pharmaceuticals, Inc. Available online: https://www.ionispharma.com/ (accessed on 10 Dec 2021).
- Lawitz, E.; Hassanein, T.; Denham, D.; Waters, M.; Borg, B.; Mille, G.; Scott, D.; Miksztal, A.; Culwell, J.; Ellis, D.; Brown, J.; Lin, W.Q. Efficacy Signals of 4-Week Oral DUR-928 in NASH Subjects. In Proceedings of the Digital International Liver Congress 2021, Virtual, 23-26 Jun 2021.