Peroxisomes are single membrane-bound organelles found in all eukaryotic cells and organisms, from yeast to plants and mammals. They are key regulators of cellular and metabolic homeostasis. These organelles play important roles in redox metabolism, the oxidation of very-long-chain fatty acids (VLCFAs), and the biosynthesis of ether phospholipids. Given the essential role of peroxisomes in cellular homeostasis, peroxisomal dysfunction has been linked to various pathological conditions, tissue functional decline, and aging. In the past few decades, a variety of cellular signaling and metabolic changes have been reported to be associated with defective peroxisomes, suggesting that many cellular processes and functions depend on peroxisomes. Peroxisomes communicate with other subcellular organelles, such as the nucleus, mitochondria, endoplasmic reticulum (ER), and lysosomes. These inter-organelle communications are highly linked to the key mechanisms by which cells surveil defective peroxisomes and mount adaptive responses to protect them from damages.
- peroxisome
- reactive oxygen species
- acetyl-CoA
- plasmalogen
- ER stress
- mitochondrial dysfunction
- apoptosis
- pexophagy
1. Introduction
2. Peroxisome Biogenesis and Peroxisomal Import Machinery
3. Key Metabolic Functions of Peroxisome
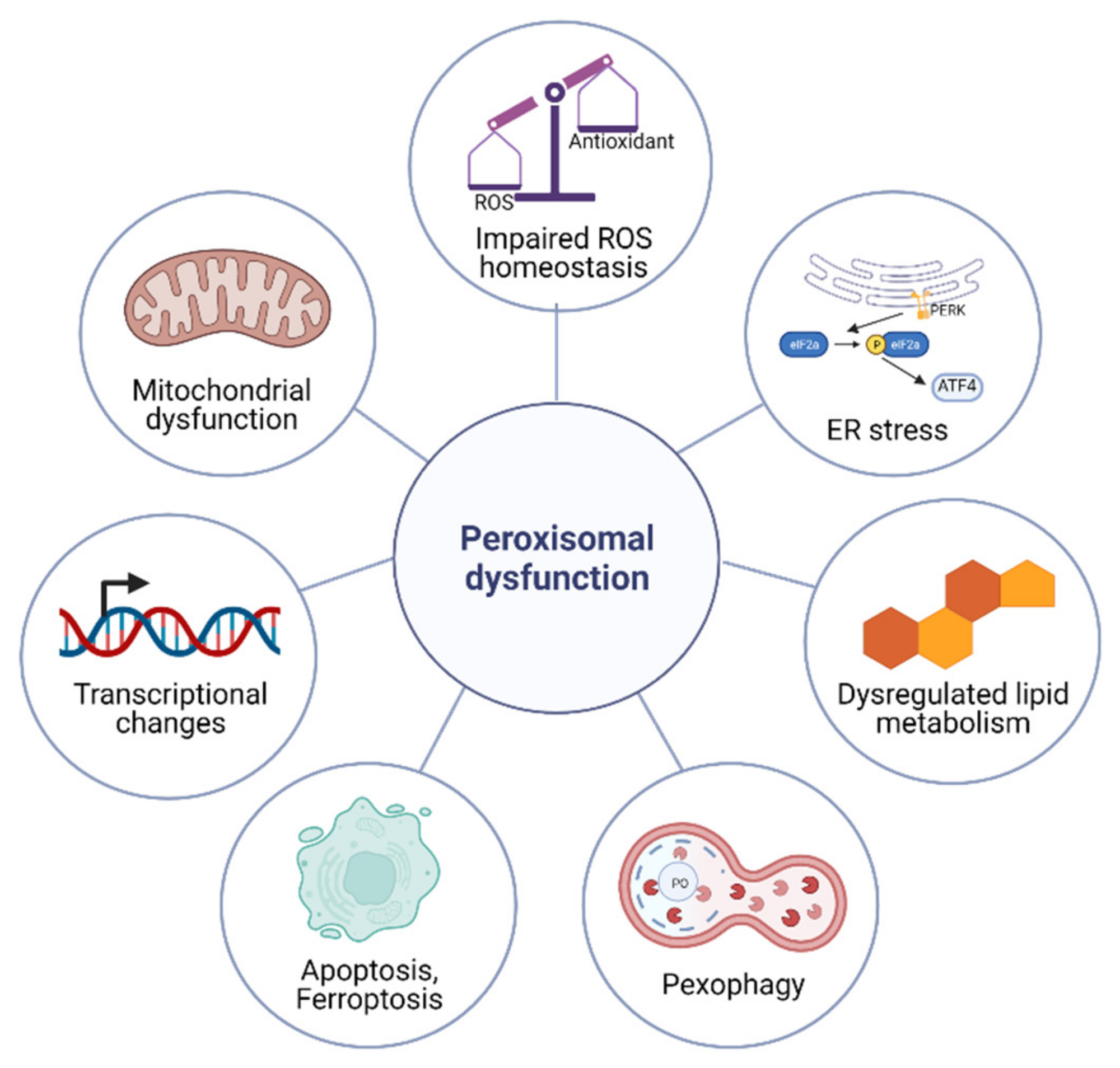
4. Cellular Responses to Peroxisomal Dysfunction
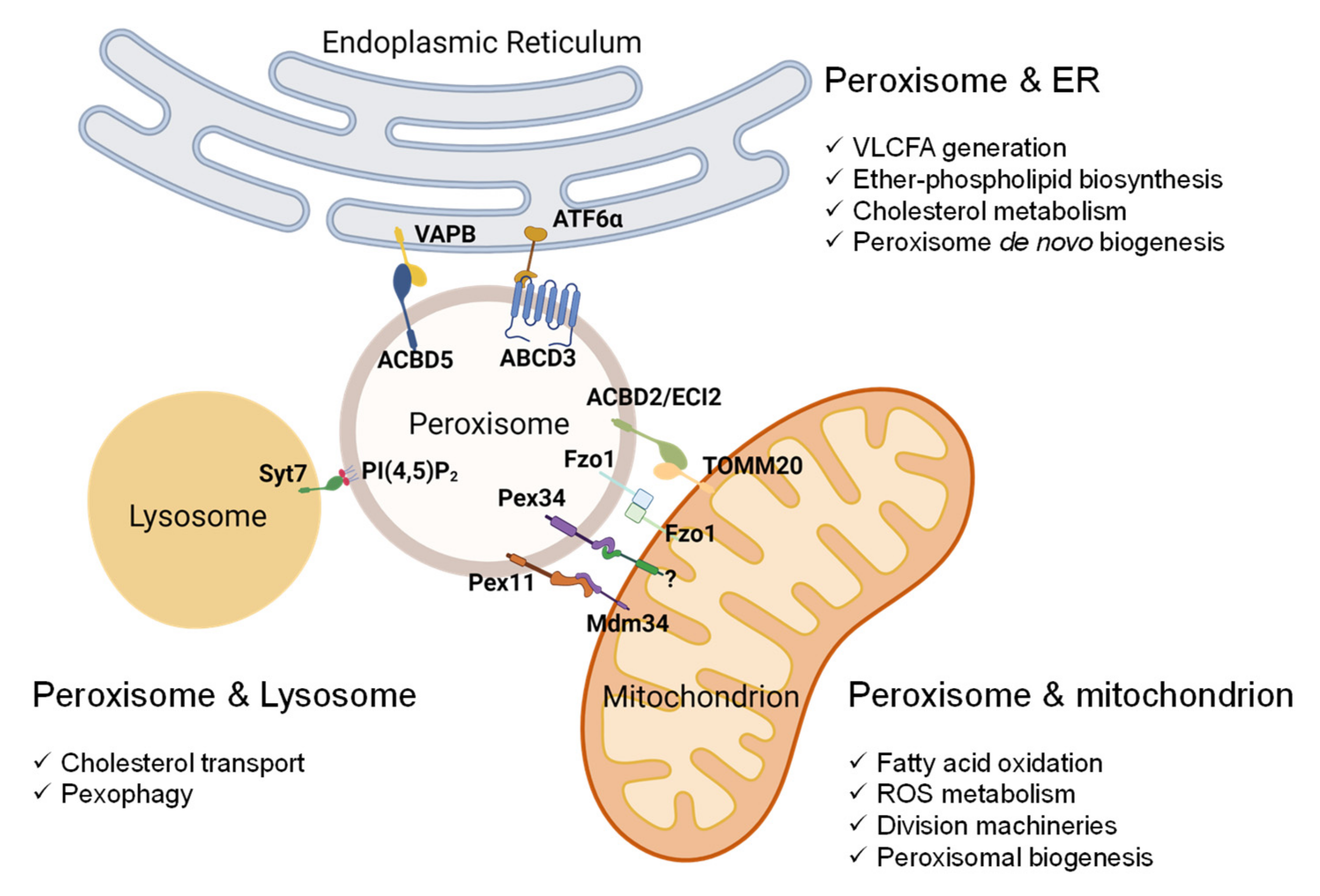
5. Peroxisome-Organelle Communication
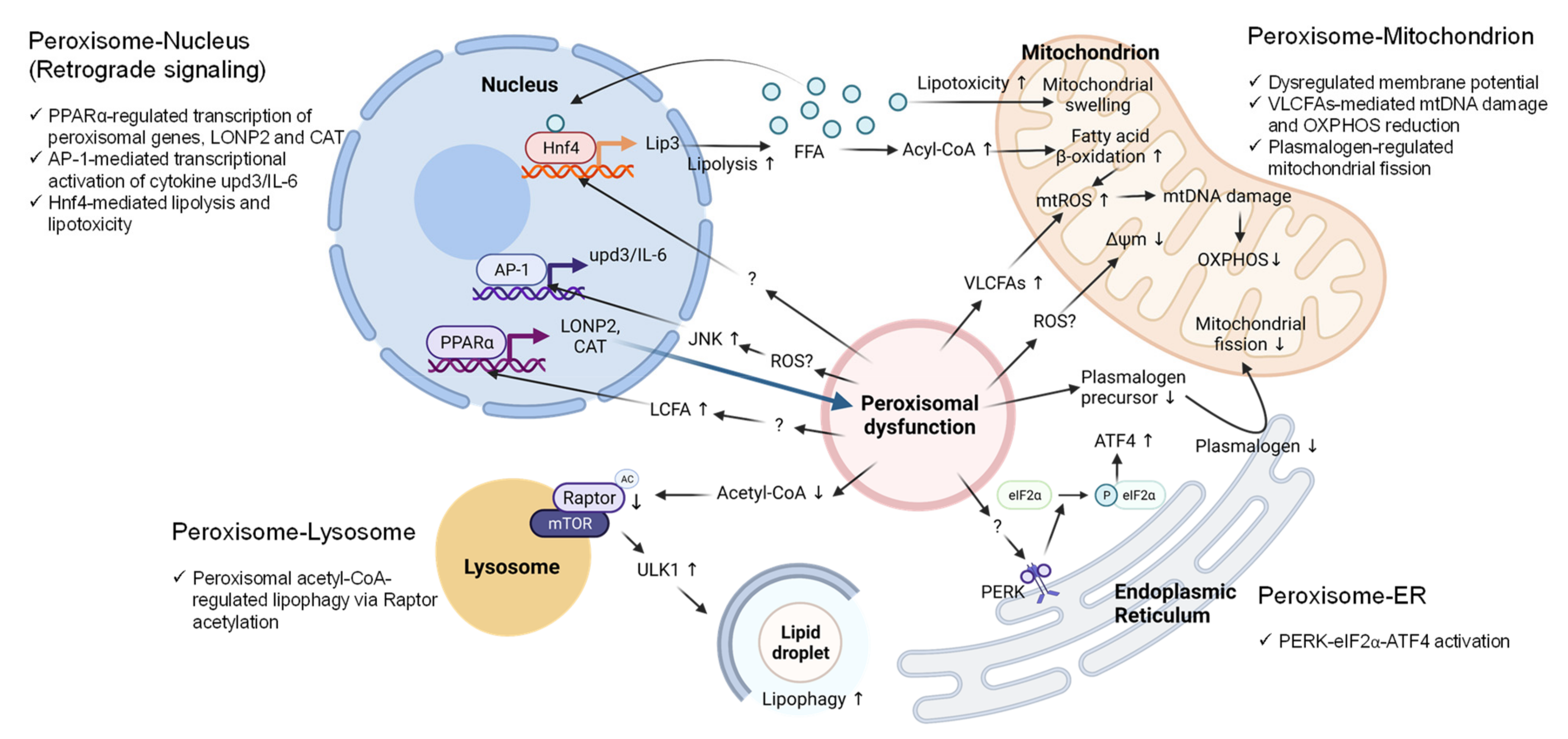
6. Peroxisomal Dysfunction in Aging and Aging-Related Diseases
This entry is adapted from the peer-reviewed paper 10.3390/antiox11020192
References
- Klouwer, F.C.; Berendse, K.; Ferdinandusse, S.; Wanders, R.J.; Engelen, M. Zellweger spectrum disorders: Clinical overview and management approach. Orphanet J. Rare. Dis. 2015, 10, 151.
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J. Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta 2016, 1863, 922–933.
- He, A.; Dean, J.M.; Lodhi, I.J. Peroxisomes as cellular adaptors to metabolic and environmental stress. Trends Cell Biol. 2021, 31, 656–670.
- Wanders, R.J. Metabolic functions of peroxisomes in health and disease. Biochimie 2014, 98, 36–44.
- Schrader, M.; Kamoshita, M.; Islinger, M. Organelle interplay-peroxisome interactions in health and disease. J. Inherit. Metab. Dis. 2020, 43, 71–89.
- Lin, T.K.; Lin, K.J.; Lin, K.L.; Liou, C.W.; Chen, S.D.; Chuang, Y.C.; Wang, P.W.; Chuang, J.H.; Wang, T.J. When Friendship Turns Sour: Effective Communication Between Mitochondria and Intracellular Organelles in Parkinson’s Disease. Front. Cell Dev. Biol. 2020, 8, 607392.
- Jin, Y.; Strunk, B.S.; Weisman, L.S. Close encounters of the lysosome-peroxisome kind. Cell 2015, 161, 197–198.
- Huang, K.; Chen, W.; Zhu, F.; Li, P.; Li, P.W.-L.; Kapahi, P.; Bai, H. RiboTag translatomic profiling of Drosophila oenocytes under aging and induced oxidative stress. BMC Genom. 2019, 20, 50.
- Huang, K.; Kim, J.; Vo, P.; Miao, T.; Bai, H. Peroxisome import stress impairs ribosome biogenesis and induces integrative stress response through eIF2α phosphorylation. bioRxiv 2021.
- Rackles, E.; Witting, M.; Forné, I.; Zhang, X.; Zacherl, J.; Schrott, S.; Fischer, C.; Ewbank, J.J.; Osman, C.; Imhof, A.; et al. Reduced peroxisomal import triggers peroxisomal retrograde signaling. Cell Rep. 2021, 34, 108653.
- Legakis, J.E.; Koepke, J.I.; Jedeszko, C.; Barlaskar, F.; Terlecky, L.J.; Edwards, H.J.; Walton, P.A.; Terlecky, S.R. Peroxisome senescence in human fibroblasts. Mol. Biol. Cell 2002, 13, 4243–4255.
- Giordano, C.R.; Terlecky, S.R. Peroxisomes, cell senescence, and rates of aging. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1358–1362.
- Narayan, V.; Ly, T.; Pourkarimi, E.; Murillo, A.B.; Gartner, A.; Lamond, A.; Kenyon, C. Deep Proteome Analysis Identifies Age-Related Processes in C. elegans. Cell Syst. 2016, 3, 144–159.
- Huang, K.; Miao, T.; Chang, K.; Kim, J.; Kang, P.; Jiang, Q.; Simmonds, A.J.; Di Cara, F.; Bai, H. Impaired peroxisomal import in Drosophila oenocytes causes cardiac dysfunction by inducing upd3 as a peroxikine. Nat. Commun. 2020, 11, 2943.
- Islinger, M.; Cardoso, M.; Schrader, M. Be different—The diversity of peroxisomes in the animal kingdom. Biochim. et Biophys. Acta 2010, 1803, 881–897.
- Huybrechts, S.J.; Van Veldhoven, P.P.; Brees, C.; Mannaerts, G.P.; Los, G.V.; Fransen, M. Peroxisome Dynamics in Cultured Mammalian Cells. Traffic 2009, 10, 1722–1733.
- Lodhi, I.J.; Semenkovich, C.F. Peroxisomes: A Nexus for Lipid Metabolism and Cellular Signaling. Cell Metab. 2014, 19, 380–392.
- Sugiura, A.; Mattie, S.; Prudent, J.; McBride, H.M. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature 2017, 542, 251–254.
- Van der Zand, A.; Gent, J.; Braakman, I.; Tabak, H.F. Biochemically distinct vesicles from the endoplasmic reticulum fuse to form peroxisomes. Cell 2012, 149, 397–409.
- Matsuzaki, T.; Fujiki, Y. The peroxisomal membrane protein import receptor Pex3p is directly transported to peroxisomes by a novel Pex19p- and Pex16p-dependent pathway. J. Cell Biol. 2008, 183, 1275–1286.
- Fang, Y.; Morrell, J.C.; Jones, J.M.; Gould, S.J. PEX3 functions as a PEX19 docking factor in the import of class I peroxisomal membrane proteins. J. Cell Biol. 2004, 164, 863–875.
- Lazarow, P.B.; Fujiki, Y. Biogenesis of Peroxisomes. Annu. Rev. Cell Biol. 1985, 1, 489–530.
- Francisco, T.; Rodrigues, T.; Dias, A.F.; Barros-Barbosa, A.; Bicho, D.; Azevedo, J.E. Protein transport into peroxisomes: Knowns and unknowns. BioEssays 2017, 39, 39.
- Platta, H.; Erdmann, R. The peroxisomal protein import machinery. FEBS Lett. 2007, 581, 2811–2819.
- Gould, S.J.; Keller, G.-A.; Hosken, N.; Wilkinson, J.; Subramani, S. A conserved tripeptide sorts proteins to peroxisomes. J. Cell Biol. 1989, 108, 1657–1664.
- Lazarow, P.B. Chapter 3.1.7. The import receptor Pex7p and the PTS2 targeting sequence. Biochim. Biophys. Acta Bioenerg. 2006, 1763, 1599–1604.
- Swinkels, B.W.; Gould, S.J.; Bodnar, A.G.; Rachubinski, R.A.; Subramani, S. A novel, cleavable peroxisomal targeting signal at the amino-terminus of the rat 3-ketoacyl-CoA thiolase. EMBO J. 1991, 10, 3255–3262.
- Meinecke, M.; Cizmowski, C.; Schliebs, W.; Krüger, V.; Beck, S.; Wagner, R.H.; Erdmann, R. The peroxisomal importomer constitutes a large and highly dynamic pore. Nat. Cell Biol. 2010, 12, 273–277.
- Rucktäschel, R.; Girzalsky, W.; Erdmann, R. Protein import machineries of peroxisomes. Biochim. Biophys. Acta Biomembr. 2011, 1808, 892–900.
- Platta, H.W.; El Magraoui, F.; Baumer, B.E.; Schlee, D.; Girzalsky, W.; Erdmann, R. Pex2 and Pex12 Function as Protein-Ubiquitin Ligases in Peroxisomal Protein Import. Mol. Cell. Biol. 2009, 29, 5505–5516.
- Platta, H.W.; Brinkmeier, R.; Reidick, C.; Galiani, S.; Clausen, M.P.; Eggeling, C. Regulation of peroxisomal matrix protein import by ubiquitination. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 838–849.
- Thoms, S.; Erdmann, R. Peroxisomal matrix protein receptor ubiquitination and recycling. Biochim. Biophys. Acta Bioenerg. 2006, 1763, 1620–1628.
- Carvalho, A.F.; Pinto, M.P.; Grou, C.P.; Alencastre, I.S.; Fransen, M.; Sa-Miranda, C.; Azevedo, J.E. Ubiquitination of Mammalian Pex5p, the Peroxisomal Import Receptor. J. Biol. Chem. 2007, 282, 31267–31272.
- Grou, C.P.; Carvalho, A.F.; Pinto, M.P.; Alencastre, I.S.; Rodrigues, T.A.; Freitas, M.O.; Francisco, T.; Sá-Miranda, C.; Azevedo, J.E. The peroxisomal protein import machinery—A case report of transient ubiquitination with a new flavor. Cell. Mol. Life Sci. 2008, 66, 254–262.
- Matsumoto, N.; Tamura, S.; Fujiki, Y. The pathogenic peroxin Pex26p recruits the Pex1p–Pex6p AAA ATPase complexes to peroxisomes. Nat. Cell Biol. 2003, 5, 454–460.
- Blok, N.B.; Tan, D.; Wang, R.Y.; Penczek, P.A.; Baker, D.; DiMaio, F.; Rapoport, T.A.; Walz, T. Unique double-ring structure of the peroxisomal Pex1/Pex6 ATPase complex revealed by cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E4017–E4025.
- Ciniawsky, S.; Grimm, I.; Saffian, D.; Girzalsky, W.; Erdmann, R.; Wendler, P. Molecular snapshots of the Pex1/6 AAA+ complex in action. Nat. Commun. 2015, 6, 7331.
- Tan, D.; Blok, N.B.; Rapoport, T.A.; Walz, T. Structures of the double-ring AAA ATPase Pex1-Pex6 involved in peroxisome biogenesis. FEBS J. 2015, 283, 986–992.
- Gardner, B.M.; Chowdhury, S.; Lander, G.C.; Martin, A. The Pex1/Pex6 complex is a heterohexameric AAA+ motor with alternating and highly coordinated subunits. J. Mol. Biol. 2015, 427, 1375–1388.
- Debelyy, M.; Platta, H.; Saffian, D.; Hensel, A.; Thoms, S.; Meyer, H.E.; Warscheid, B.; Girzalsky, W.; Erdmann, R. Ubp15p, a Ubiquitin Hydrolase Associated with the Peroxisomal Export Machinery. J. Biol. Chem. 2011, 286, 28223–28234.
- Grou, C.P.; Francisco, T.; Rodrigues, T.A.; Freitas, M.O.; Pinto, M.P.; Carvalho, A.F.; Domingues, P.; Wood, S.A.; Rodríguez-Borges, J.E.; Sá-Miranda, C.; et al. Identification of Ubiquitin-specific Protease 9X (USP9X) as a Deubiquitinase Acting on Ubiquitin-Peroxin 5 (PEX5) Thioester Conjugate. J. Biol. Chem. 2012, 287, 12815–12827.
- Schrader, M.; Reuber, B.E.; Morrell, J.C.; Jimenez-Sanchez, G.; Obie, C.; Stroh, T.A.; Valle, D.; Schroer, T.A.; Gould, S.J. Expression of PEX11beta mediates peroxisome proliferation in the absence of extracellular stimuli. J. Biol. Chem. 1998, 273, 29607–29614.
- Schrader, M.; Fahimi, H.D. Growth and Division of Peroxisomes. Adv. Appl. Microbiol. 2006, 255, 237–290.
- Fagarasanu, A.; Fagarasanu, M.; Rachubinski, R.A. Maintaining peroxisome populations: A story of division and inheritance. Annu. Rev. Cell Dev. Biol. 2007, 23, 321–344.
- Thoms, S.; Erdmann, R. Dynamin-related proteins and Pex11 proteins in peroxisome division and proliferation. FEBS J. 2005, 272, 5169–5181.
- Okumoto, K.; Tamura, S.; Honsho, M.; Fujiki, Y. Peroxisome: Metabolic Functions and Biogenesis. Adv. Exp. Med. Biol. 2020, 1299, 3–17.
- Ebberink, M.S.; Koster, J.; Visser, G.; van Spronsen, F.; Stolte-Dijkstra, I.; Smit, G.P.; Fock, J.M.; Kemp, S.; Wanders, R.J.; Waterham, H.R. A novel defect of peroxisome division due to a homozygous non-sense mutation in the PEX11β gene. J. Med. Genet. 2012, 49, 307–313.
- Itoyama, A.; Honsho, M.; Abe, Y.; Moser, A.; Yoshida, Y.; Fujiki, Y. Docosahexaenoic acid mediates peroxisomal elongation, a prerequisite for peroxisome division. J. Cell Sci. 2012, 125, 589–602.
- Kobayashi, S.; Tanaka, A.; Fujiki, Y. Fis1, DLP1, and Pex11p coordinately regulate peroxisome morphogenesis. Exp. Cell Res. 2007, 313, 1675–1686.
- Opaliński, Ł.; Kiel, J.A.; Williams, C.; Veenhuis, M.; Van Der Klei, I.J. Membrane curvature during peroxisome fission requires Pex11. EMBO J. 2011, 30, 5–16.
- Yoshida, Y.; Niwa, H.; Honsho, M.; Itoyama, A.; Fujiki, Y. Pex11mediates peroxisomal proliferation by promoting deformation of the lipid membrane. Biol. Open 2015, 4, 710–721.
- Su, J.; Thomas, A.S.; Grabietz, T.; Landgraf, C.; Volkmer, R.; Marrink, S.J.; Williams, C.; Melo, M.N. The N-terminal amphipathic helix of Pex11p self-interacts to induce membrane remodelling during peroxisome fission. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1292–1300.
- Itoyama, A.; Michiyuki, S.; Honsho, M.; Yamamoto, T.; Moser, A.; Yoshida, Y.; Fujiki, Y. Mff functions with Pex11pβ and DLP1 in peroxisomal fission. Biol. Open 2013, 2, 998–1006.
- Bonekamp, N.A.; Völkl, A.; Fahimi, H.D.; Schrader, M. Reactive oxygen species and peroxisomes: Struggling for balance. BioFactors 2009, 35, 346–355.
- Islinger, M.; Li, K.W.; Seitz, J.; Völkl, A.; Lüers, G.H. Hitchhiking of Cu/Zn Superoxide Dismutase to Peroxisomes–Evidence for a Natural Piggyback Import Mechanism in Mammals. Traffic 2009, 10, 1711–1721.
- del Río, L.A.; Corpas, F.J.; Sandalio, L.M.; Palma, J.M.; Gómez, M.; Barroso, J.B. Reactive oxygen species, antioxidant systems and nitric oxide in peroxisomes. J. Exp. Bot. 2002, 53, 1255–1272.
- Wanders, R.J.; Waterham, H.R. Biochemistry of Mammalian Peroxisomes Revisited. Annu. Rev. Biochem. 2006, 75, 295–332.
- Cipolla, C.M.; Lodhi, I.J. Peroxisomal Dysfunction in Age-Related Diseases. Trends Endocrinol. Metab. 2017, 28, 297–308.
- Wanders, R.J.A.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2016, 3, 83.
- Nagan, N.; Zoeller, R.A. Plasmalogens: Biosynthesis and functions. Prog. Lipid Res. 2001, 40, 199–229.
- Dean, J.M.; Lodhi, I.J. Structural and functional roles of ether lipids. Protein Cell 2018, 9, 196–206.
- Honsho, M.; Tanaka, M.; Zoeller, R.A.; Fujiki, Y. Distinct Functions of Acyl/Alkyl Dihydroxyacetonephosphate Reductase in Peroxisomes and Endoplasmic Reticulum. Front. Cell Dev. Biol. 2020, 8, 855.
- Gallego-García, A.; Monera-Girona, A.J.; Pajares-Martínez, E.; Bastida-Martínez, E.; Pérez-Castaño, R.; Iniesta, A.A.; Fontes, M.; Padmanabhan, S.; Elías-Arnanz, M. A bacterial light response reveals an orphan desaturase for human plasmalogen synthesis. Science 2019, 366, 128–132.
- Werner, E.R.; Keller, M.A.; Sailer, S.; Lackner, K.; Koch, J.; Hermann, M.; Coassin, S.; Golderer, G.; Werner-Felmayer, G.; Zo-eller, R.A.; et al. The TMEM189 gene encodes plasmanylethanolamine desaturase which introduces the characteristic vinyl ether double bond into plasmalogens. Proc. Natl. Acad. Sci. USA 2020, 117, 7792–7798.
- De Duve, C.; Baudhuin, P. Peroxisomes (microbodies and related particles). Physiol. Rev. 1966, 46, 323–357.
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630.
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. et Biophys. Acta Mol. Basis Dis. 2011, 1822, 1363–1373.
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18, 1126.
- Sebastiani, P.; Federico, A.; Morris, M.; Gurinovich, A.; Tanaka, T.; Chandler, K.B.; Andersen, S.L.; Denis, G.; Costello, C.E.; Ferrucci, L.; et al. Protein signatures of centenarians and their offspring suggest centenarians age slower than other humans. Aging Cell 2021, 20, e13290.
- Titorenko, V.I.; Terlecky, S.R. Peroxisome Metabolism and Cellular Aging. Traffic 2010, 12, 252–259.
- Lizard, G.; Rouaud, O.; Demarquoy, J.; Cherkaoui-Malki, M.; Iuliano, L. Potential roles of peroxisomes in Alzheimer’s disease and in dementia of the Alzheimer’s type. J. Alzheimer Dis. 2012, 29, 241–254.
- Jo, D.S.; Park, N.Y.; Cho, D.-H. Peroxisome quality control and dysregulated lipid metabolism in neurodegenerative diseases. Exp. Mol. Med. 2020, 52, 1486–1495.
- Pharaoh, G.; Sataranatarajan, K.; Street, K.; Hill, S.; Gregston, J.; Ahn, B.; Kinter, C.; Kinter, M.; Van Remmen, H. Metabolic and Stress Response Changes Precede Disease Onset in the Spinal Cord of Mutant SOD1 ALS Mice. Front. Neurosci. 2019, 13, 487.
- Qin, F.; Lennon-Edwards, S.; Lancel, S.; Biolo, A.; Siwik, D.A.; Pimentel, D.R.; Dorn, G.W.; Kang, Y.J.; Colucci, W.S. Cardiac-specific overexpression of catalase identifies hydrogen peroxide-dependent and -independent phases of myocardial remodeling and prevents the progression to overt heart failure in G(alpha)q-overexpressing transgenic mice. Circ. Heart Fail. 2010, 3, 306–313.
- Piao, L.; Dorotea, D.; Jiang, S.; Koh, E.H.; Oh, G.T.; Ha, H. Impaired Peroxisomal Fitness in Obese Mice, a Vicious Cycle Exacerbating Adipocyte Dysfunction via Oxidative Stress. Antioxid. Redox Signal. 2019, 31, 1339–1351.
- Hwang, I.; Lee, J.; Huh, J.Y.; Park, J.; Lee, H.B.; Ho, Y.-S.; Ha, H. Catalase Deficiency Accelerates Diabetic Renal Injury Through Peroxisomal Dysfunction. Diabetes 2012, 61, 728–738.
- Francque, S.; Szabo, G.; Abdelmalek, M.F.; Byrne, C.D.; Cusi, K.; Dufour, J.-F.; Roden, M.; Sacks, F.; Tacke, F. Nonalcoholic steatohepatitis: The role of peroxisome proliferator-activated receptors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 24–39.
- Marcus, D.L.; Thomas, C.; Rodriguez, C.; Simberkoff, K.; Tsai, J.S.; Strafaci, J.A.; Freedman, M.L. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp. Neurol. 1998, 150, 40–44.
- Sheikh, F.G.; Pahan, K.; Khan, M.; Barbosa, E.; Singh, I. Abnormality in catalase import into peroxisomes leads to severe neurological disorder. Proc. Natl. Acad. Sci. USA 1998, 95, 2961–2966.
- Goodenowe, D.B.; Cook, L.L.; Liu, J.; Lu, Y.; Jayasinghe, D.A.; Ahiahonu, P.W.; Heath, D.; Yamazaki, Y.; Flax, J.; Krenitsky, K.F.; et al. Peripheral ethanolamine plasmalogen deficiency: A logical causative factor in Alzheimer’s disease and dementia. J. Lipid Res. 2007, 48, 2485–2498.
- Graham, W.V.; Bonito-Oliva, A.; Sakmar, T.P. Update on Alzheimer’s Disease Therapy and Prevention Strategies. Annu. Rev. Med. 2017, 68, 413–430.
- Cimini, A.; Moreno, S.; D’Amelio, M.; Cristiano, L.; D’Angelo, B.; Falone, S.; Benedetti, E.; Carrara, P.; Fanelli, F.; Cecconi, F.; et al. Early biochemical and morphological modifications in the brain of a transgenic mouse model of Alzheimer’s disease: A role for peroxisomes. J. Alzheimer Dis. 2009, 18, 935–952.
- Fanelli, F.; Sepe, S.; D’Amelio, M.; Bernardi, C.; Cristiano, L.; Cimini, A.; Cecconi, F.; Ceru’, M.P.; Moreno, S. Age-dependent roles of peroxisomes in the hippocampus of a transgenic mouse model of Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 8.
- Nunomura, A.; Tamaoki, T.; Motohashi, N.; Nakamura, M.; McKeel, D.W., Jr.; Tabaton, M.; Lee, H.-G.; Smith, M.A.; Perry, G.; Zhu, X. The Earliest Stage of Cognitive Impairment in Transition from Normal Aging to Alzheimer Disease Is Marked by Prominent RNA Oxidation in Vulnerable Neurons. J. Neuropathol. Exp. Neurol. 2012, 71, 233–241.
- Kou, J.; Kovacs, G.G.; Höftberger, R.; Kulik, W.; Brodde, A.; Forss-Petter, S.; Hönigschnabl, S.; Gleiss, A.; Brügger, B.; Wanders, R.; et al. Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathol. 2011, 122, 271–283.
- Astarita, G.; Jung, K.M.; Berchtold, N.C.; Nguyen, V.Q.; Gillen, D.L.; Head, E.; Cotman, C.W.; Piomelli, D. Deficient liver biosynthesis of docosahexaenoic acid correlates with cognitive impairment in Alzheimer’s disease. PLoS ONE 2010, 5, e12538.
- Shi, R.; Zhang, Y.; Shi, Y.; Shi, S.; Jiang, L. Inhibition of peroxisomal β-oxidation by thioridazine increases the amount of VLCFAs and Aβ generation in the rat brain. Neurosci. Lett. 2012, 528, 6–10.
- Inestrosa, N.C.; Carvajal, F.J.; Zolezzi, J.M.; Tapia-Rojas, C.; Serrano, F.; Karmelic, D.; Toledo, E.M.; Toro, A.; Toro, J.; Santos, M.J. Peroxisome proliferators reduce spatial memory impairment, synaptic failure, and neurodegeneration in brains of a double transgenic mice model of Alzheimer’s disease. J. Alzheimer Dis. 2013, 33, 941–959.
- Berger, J.; Dorninger, F.; Forss-Petter, S.; Kunze, M. Peroxisomes in brain development and function. Biochim. Biophys. Acta 2016, 1863, 934–955.
- Dai, D.-F.; Chen, T.; Johnson, S.; Szeto, H.; Rabinovitch, P.S. Cardiac Aging: From Molecular Mechanisms to Significance in Human Health and Disease. Antioxid. Redox Signal. 2012, 16, 1492–1526.
- Yazdanyar, A.; Newman, A.B. The Burden of Cardiovascular Disease in the Elderly: Morbidity, Mortality, and Costs. Clin. Geriatr. Med. 2009, 25, 563–577.
- Rodgers, J.L.; Jones, J.; Bolleddu, S.I.; Vanthenapalli, S.; Rodgers, L.E.; Shah, K.; Karia, K.; Panguluri, S.K. Cardiovascular Risks Associated with Gender and Aging. J. Cardiovasc. Dev. Dis. 2019, 6, 19.
- Mittler, R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 2002, 7, 405–410.
- Bäumer, A.T.; Flesch, M.; Wang, X.; Shen, Q.; Feuerstein, G.Z.; Böhm, M. Antioxidative Enzymes in Human Hearts with Idiopathic Dilated Cardiomyopathy. J. Mol. Cell. Cardiol. 2000, 32, 121–130.
- Colasante, C.; Chen, J.; Ahlemeyer, B.; Baumgart-Vogt, E. Peroxisomes in cardiomyocytes and the peroxisome/peroxisome proliferator-activated receptor-loop. Thromb. Haemost. 2015, 113, 452–463.
- Koh, J.T.; Choi, H.H.; Ahn, K.Y.; Kim, J.U.; Kim, J.H.; Chun, J.-Y.; Baik, Y.H.; Kim, K.K. Cardiac Characteristics of Transgenic Mice Overexpressing Refsum Disease Gene-Associated Protein within the Heart. Biochem. Biophys. Res. Commun. 2001, 286, 1107–1116.
- Wanders, R.J.; Komen, J.C. Peroxisomes, Refsum’s disease and the alpha- and omega-oxidation of phytanic acid. Biochem. Soc. Trans. 2007, 35, 865–869.
- Miard, S.; Picard, F. Obesity and aging have divergent genomic fingerprints. Int. J. Obes. 2008, 32, 1873–1874.
- Barzilai, N.; Huffman, D.M.; Muzumdar, R.H.; Bartke, A. The Critical Role of Metabolic Pathways in Aging. Diabetes 2012, 61, 1315–1322.
- Jura, M.; Kozak, L. Obesity and related consequences to ageing. AGE 2016, 38, 23.
- Goth, L.; Eaton, J.W. Hereditary catalase deficiencies and increased risk of diabetes. Lancet 2000, 356, 1820–1821.