1. Introduction
Coronaviruses (CoVs) are RNA viruses with a single strand that belong to the Coronaviridae family while four CoVs categories have thus far been identified: α, β, γ, δ. SARS-CoV2 penetrates human cells by attaching to the angiotensin-converting enzyme 2 (ACE2), abundant in alveolar lung cells, vascular endothelium, cardiac myocytes, and other cells [
1]. The novel coronavirus disease 2019 (COVID-19) emerged as a severe acute respiratory illness and was proclaimed a pandemic on 30 January 2020, affecting primarily the residents of Wuhan, Hubei Province in China [
2,
3].
The course of the disease is mild in the large proportion of patients, while severe cases with hospitalization and high mortality rates also occur. Physicians have tried to classify the disease course by dividing it into four stages [
4,
5]. In the first stage (Stage I), fever, dry cough, tiredness, and myalgia are the most common symptoms which are, however, not specific to the disease. In the second stage (Stage II), bilateral pulmonary parenchymal ground-glass and consolidative pulmonary opacities are presented on the computed tomography scan in the vast majority of COVID-19 patients with viral pneumonia [
4]. A hypercoagulable state has been observed, especially in hospitalized patients in Stage III of the disease, driven by abnormal coagulation cascades’ activation [
5]. Finally, multiorgan failure on top of excessive hypoxemia appears in Stage IV with hyperresponsiveness of the immune system [
4,
5]. This stage is characterized by rapid elevation of inflammatory circulating cytokines such as interleukin (IL)-1, IL-2, IL-6, and IL-7, tumor necrosis factor (TNF)-α, granulocyte–macrophage colony-stimulating factor (GM–CSF), macrophage inflammatory protein 1-α (MIP-1α), C-reactive protein (CRP), ferritin, and D-dimer [
6]. This extreme inflammatory response causes severe adult Acute Respiratory Distress Syndrome (ARDS) and the so-called “cytokine storm” [
6].
As such, the mitigation of the excessive inflammatory immune response is of high scientific interest and clinical relevance. Recently, anti-inflammatory drugs and immunomodulators have been studied as potential therapies to minimize cardiovascular and systemic adverse effects.
A challenge for physicians is the optimal management of an abnormal hyper-inflammatory state with elevated pro- and inflammatory cytokines, which could drive ARDS and has been described in numerous hospitalized COVID-19 patients [
7]. To the best of our knowledge, a similar disorder has been described in juvenile Still disease, revealed as a cytokine storm leading to macrophage activation syndrome (MAS), secondary haemophagocytic lymphohistiocytosis (sHLH), or cytokine release syndrome (CRS) [
6,
20]. These pathological entities are more commonly driven by viral infections, autoimmune disorders, malignancy (HLH and MAS), sepsis, and the administration of chimeric antigen receptor T cell therapy (CRS) [
21,
22]. It is characterized by sudden fever, respiratory and kidney failure, hypotensive shock, and diffuse coagulation disorders, while laboratory examinations reveal anemia, neutrophilia, thrombocytopenia, and marked lymphopenia [
23,
24]. Multi-organ failure presented in many cases, while four molecular cascades were responsible for its course—complement, kinin, clotting, and fibrinolysis systems. A similar phenomenon has been observed in patients with severe COVID-19 pneumonia with primary HLH as the potential underlying cause [
20,
25].
The assumed underlying mechanism is the rocket of secreted inflammatory cytokine levels in the bloodstream of the patients. The critical pathogenic cytokines appear to vary according to illness, with IL-1β having an orchestrating role in Still disease, IL-18 in MAS, and IL-6 in CRS [
26]. As far as the severe COVID-19 disease is concerned, the widespread hypothesis is that, in the early phase, failure of perforin, natural killer cells (NK), and CD8+ cytotoxic T-cells leads to cell lysis, initiating apoptosis of virally infected cells while interferon-γ (IFN-γ) causes excessive macrophage activation [
25,
27]. Multiple studies have revealed that the toll-like receptors (TLRs) and activated inflammasomes (Caspases) first release the primarily inflammatory component of the disease, IL-1β [
28,
29]. Parallel delayed secretion of type Ⅰ and Ⅲ IFNs, including IFN α/ß, in the early phase of infection and excessive secretion of pro-inflammatory cytokines from mononuclear macrophages is described in the later stage [
30]. The cells release modest levels of antiviral factors—IFNs—as well as high amounts of pro-inflammatory cytokines—IL-1, IL-6, and TNF—and particular chemokines—C-C pattern chemokine ligand (CCL)-2, CCL-3, and CCL-5 [
31,
32]. Linear association of disease severity course and the type of elevated cytokines has not been well established yet. Several studies suggest that higher levels of IL-1β, IL-1RA, IL-7, IL-8, IL-10, IFN-ɣ, MCP-1, MIP-1α, G-CSF, and TNF-α have been observed in severe infection with marginal statistical significance [
27,
33]. Airway and alveolar epithelial cell apoptosis was induced by IFN-αβ and IFN-γ, increasing the inflammatory cell infiltration. Apoptosis of endothelial and epithelial cells affects the pulmonary microvascular and alveolar epithelial cell barriers, causing vascular leakage, alveolar edema, and, eventually, hypoxia [
7,
25]. The studies mentioned above emphasize that a failure in initial type-Ⅰ and Ⅲ IFN responses to SARS-CoV-2 leads to an excessive late immune response and severe form of COVID-19. The pro-inflammatory feed-forward loop of cytokines on innate immune cells results in a cytokine storm, coagulopathy, and acute respiratory distress syndrome (ARDS) [
34,
35]. On the other hand, in contrast with the widespread hypothesis of failure of the immune system, two other studies presented an interesting and quite different concept of cytokine storm onset, differentiating it into two stages. In the first, a short-term immune-deficient state is considered, followed by a second overactive immune condition which tries to counterbalance the agitated entropy from temporary immune target failure, driving to a cytokine storm [
7,
36]. As such, further molecular research on patients presented with cytokine storm caused by COVID-19 is required to clarify the phenomenon (
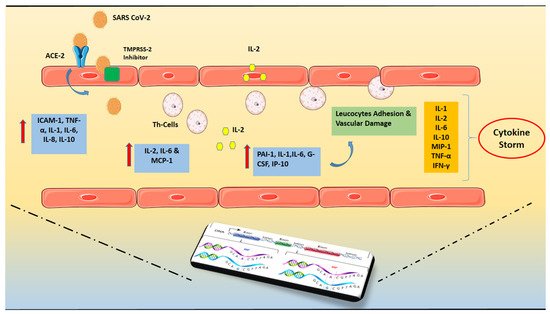
Figure 1).
Figure 1. SARS-CoV-2 invasion and hyper-inflammatory state in close relation with genetic predisposition. The presence of Angiotensin-converting enzyme 2 (ACE2) and Transmembrane protease serine 2 (TMPRSS-2) that may cleave the viral spike is required for SARS-CoV-2’s cell invasion. Increased levels of pro-inflammatory cytokines, particularly the soluble interleukin 2-receptor (IL-2R) and interleukin-6 (IL-6) have been found. Soluble IL-2R (sIL-2R) is mostly released by activated T helper lymphocytes, although it may also be secreted by endothelial cells (ECs). The capillary leak is caused by the binding of IL-6 and IL-2 to their receptors. The persistent burdening of the endothelium results in increased release of inflammatory cytokines and immune system overreaction, resulting in the so-called “cytokine storm”. The above mentioned hyper-inflammatory state is in close relation with the individual genetic profile which can potentially govern the course of the disease. Abbreviations: SARS CoV-2 = Severe acute respiratory syndrome Coronavirus-2, ACE2 = Angiotensin-converting enzyme 2, TMPRSS = Transmembrane protease serine 2, IL = Interleukin, ΡAΙ-1 = Plasminogen activator inhibitor-1, TNF = Tumor Necrosis Factor, ICAM = Intercellular Adhesion Molecule 1, MCP-1 = monocyte chemoattractant protein-1, G-CSF = Granulocyte colony-stimulating factor, IP-10 = Interferon gamma-induced protein 10, MIP-1 = Macrophage inflammatory protein-1, IFN = Interferon.