Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Organic
Schiff bases are a vast group of compounds characterized by the presence of a double bond linking carbon and nitrogen atoms, the versatility of which is generated in the many ways to combine a variety of alkyl or aryl substituents.
- Dihydrazides
- Hydrazones
- Hydrazide
1. Dihydrazides
1.1. Structure
The plurality of dihydrazide compounds (Figure 1), similar to that of hydrazide–hydrazones, results from the variety of alkyl or aryl fragments and their substituents that can be linked by a diamide bridge. The symmetrical structure determines the nomenclature: each time, researchers start naming with the carbonyl group, invariably obtaining a monosubstituted hydrazide. Thus, including both reading possibilities, the name mentioned above can be adopted.
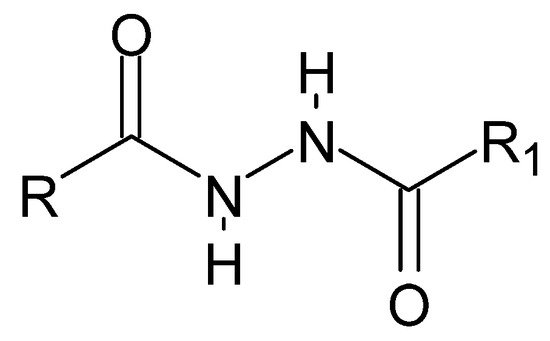
Figure 1. The basic structure of dihydrazides, where R and R1 is an alkyl, aryl, or hydrogen group.
The synthesis of dihydrazides—methanoic and ethanoic acid dihydrazides—was first described by T. Curtis, N. Schwann and G. Schöfer in 1895 [100].
1.2. Importance
Dihydrazides are important as antibacterial, antifungal and antiparasitic agents. The fact that it is the hydrazide moiety, which is the key to the biological activity of similar compounds, was already reported in the research by Raymond Cavier and Richard Rips in 1965, in which they observed that the replacement of the diisopropylidenemalonyl hydrazide moiety with an amide moiety reduces the compound’s activity against the nematode S. obvelata [114].
An example of another property of some dihydrazides is chemiluminescence, i.e., the emission of radiation manifested by the light effect (ultraviolet, visible light and infrared), which results from the recombination of the electron–hole pair in an electronically excited product or an intermediate product, resulting from the action of an external factor, which in this case is a chemical reaction, on a given compound [115]. One of the earliest described chemiluminophores—as early as 1928 [116], commonly called luminol, is 3-aminophthalic acid hydrazide, with the capacity to emit light at λ = 425 nm (blue light) [92]. The exchange of the carbonyl groups with the secondary amino groups while maintaining the symmetry (amide groups connected through the carbon atoms of carbonyl groups) causes the compounds to lose their chemiluminescent properties. They are also negatively affected by the presence of electron-withdrawing substituents (-Cl, -NO2), while electron-donating substituents (-NH2, -OH) strengthen them [92]. The chemiluminescent properties allow the use of dihydrazides, as they are highly sensitive to pH changes and are indicators of the endpoint of acid–base titration (N′-formyl-rhodamine B dihydrazide) [117], as DNA probes and in immunoassays (luminol) [118].
The presence of the amide-analogous -C(=O)-NH-NH- group enables the occurrence of the aforementioned amide-iminol tautomerism, presented below (Figure 2 and Figure 3) [92].
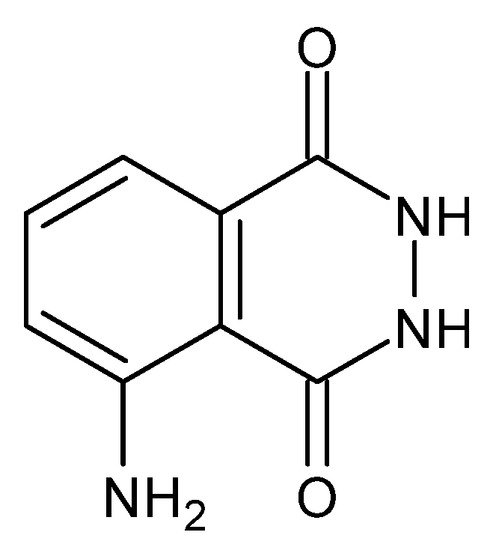
Figure 2. Structure of 3-aminophthalic acid hydrazide (luminol) [92].
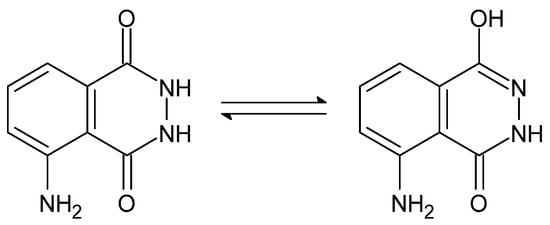
Figure 3. Amide-iminol tautomerism of luminol [92].
Dihydrazides are also used as catalysts in highly enantioselective reactions leading to the formation of asymmetric aldols [83].
2. Hydrazones
2.1. Structure
A special type of hydrazides are hydrazones, the main distinguishing feature of which is the presence of an imine bond in their moiety (Figure 4), which in turn participates in imine–enamine tautomerization (Figure 5) due to the presence of the α-hydrogen atom.
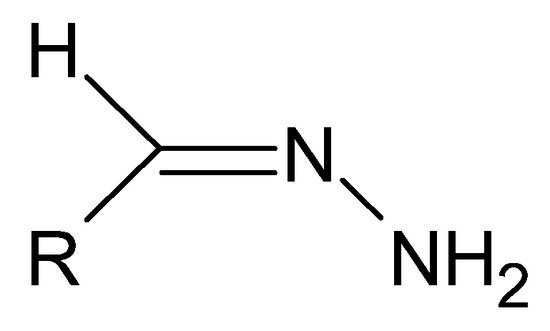
Figure 4. The basic structure of hydrazones, where R is an alkyl or aryl group.
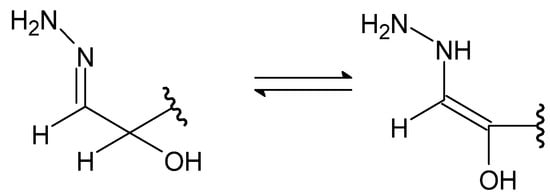
Figure 5. The imine–enamine tautomerization of hydrazones.
Characteristic signals are visible in the 1H NMR spectrum in the form of a singlet with a chemical shift of δ 8.16–8.67 ppm for -CH= and a singlet with a chemical shift of δ 10.45 12.25 ppm for -NH- [119].
The N-N bond can be reduced to the -NH2 group [120] or the whole hydrazone molecule can be reduced to a hydrazide by reductive acylation [121]. The C=N bond is susceptible to a nucleophilic attack. It can be hydrolyzed, oxidized or reduced—it willingly restores the C=O carbonyl group. It can undergo the nucleophilic addition of an organometallic compound (containing Li, Mg, Ce and Yb atoms), as well as of the intramolecular group -SH [107,122]. Under appropriate conditions, hydrazones can react with α,β-unsaturated aldehydes, giving interesting dihydrazide connections with a lactam ring [123].
2.2. Importance
Hydrazones have uses, among others, as pesticides, insecticides, nematicides, rodenticides or plant-growth regulators [124].
Sulfonyl hydrazones are important as antidepressants, analgesics, anti-inflammatory, anti-cancer, antifungal, and antibacterial agents—they can act as strong inhibitors of tyrosine phosphatase B (PtpB) of bacteria from the M. tuberculosis strain [125] and are antidiabetic [126]. This particular type of hydrazone is often tested for antimicrobial properties alongside its analogues—sulfonamides [125]. Equally common is the use of aryl hydrazones, in which the hydrazone moiety is directly linked to the aromatic ring, in medicine [127].
As with Schiff bases, hydrazones are considered multidentate ligands for their chelating capacity. Complexes of hydrazones with transition metal ions may show increased antimicrobial activity compared to uncomplexed ligands, where it is the metal ion that has the capacity to disturb cellular activity, while the ligand “assists”, increasing the lipophilicity of the ion’s environment, thus allowing for its penetration of the cell membrane [61,128,129,130].
Less frequently, the opposite phenomenon occurs—a decrease in the antibacterial activity of the sulfonyl hydrazones and their complexes (Figure 6) in relation to their ligands, which is explained by the fact that the electron density is reduced on the donor (O, N) atoms involved in the formation of coordination bonds with the tran-sition metal ion in the complex [131].
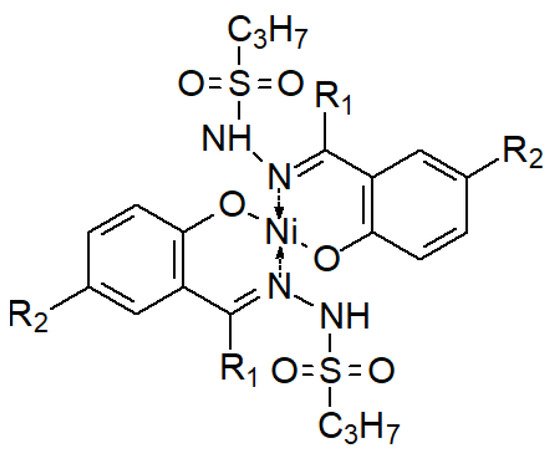
Figure 6. A complex of two hydrazone ligands and one nickel(II) ion [131].
2.3. Classification
Among hydrazones, as well as among hydrazides, besides carbonyl, one can distinguish sulfonyl hydrazones. However, the sulfonyl group does not replace the nitrogen–carbon bond inherent to hydrazones, but is attached to the amino group, which is terminal in classic hydrazones (Figure 7 below).
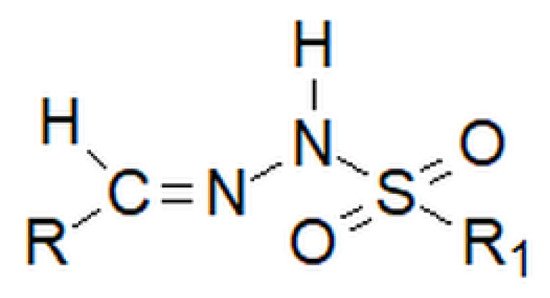
Figure 7. Basic structure of the sulfonyl hydrazone, where R and R1 are alkyl or aryl groups.
The oxidation of thiol leads to sulfonyl acid, and with chlorinating reagent (SOCl2, POCl3, PCl5 and cyanuric acid chloride)—to sulfonyl acid chloride, which can further react with hydrazine and aldehyde or ketone to give sulfonyl hydrazone [126]. A special type of sulfonyl hydrazone is 4-toluenesulfonylhydrazone, obtained by reacting an aldehyde or ketone with tosylhydrazide, which is the product of the reaction of tosyl chloride and hydrazine [132].
3. Hydrazide–Hydrazones
3.1. Structure
Hydrazide–hydrazones are a numerous group of hybrid molecules which can connect diametrically different alkyl or aryl fragments through an unsymmetrical amide–imine bridge -C(=O)-NH-N=CH-, which builds the heterogeneity of this group of compounds (Figure 29). Its name unsurprisingly comes from the dual possibility of reading the order of atoms in a moiety. If the nomenclature begins with the carbonyl group, it can be said that researchers are dealing with monosubstituted carbonyl hydrazides, while if researchers regard the imine bond as the beginning, the—monosubstituted—hydrazone clearly appears.
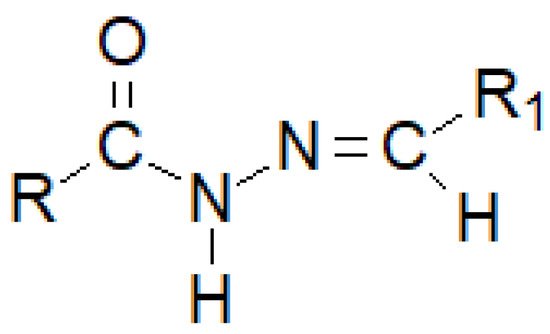
Figure 8. The basic structure of a hydrazide–hydrazone, where R and R1 are either alkyl or an aryl groups.
The hydrazide–hydrazone moiety can be identified via spectroscopic methods. The IR spectrum shows signals at around 3050 cm−1, 1650 cm−1 and 1550 cm−1, corresponding to the -NH-, C=O and C=N group, respectively. Two singlets appear in the 1H NMR spectrum, one in the range of δ 8–9 ppm and the other in the range of δ 10–13 ppm, signals corresponding to the -CH= and -NH- group, respectively. In the 13C NMR spectrum, there are signals of carbon atoms from the CH= and C=O group in the range of δ 145–160 ppm and δ 160–170 ppm, respectively [133].
3.2. Importance
Hydrazide–hydrazones are synthesized in search of effective antibacterial and antifungal agents—especially due to the growing problem of antibiotic resistance [134]. In many cases, it is the presence of electron-withdrawing aromatic ring substituents that is essential for the antimicrobial activity of hydrazide–hydrazones. Studies on the biological activity of, among others, the following compound (Figure 30) provide an example of the mentioned relationship. The lowest MIC values (μg/mL) are shown for the chlorine substituent in position 2, against E. coli, S. aureus and B. subtilis, 0.31, 0.62 and 0.31, respectively. They were compared with the MIC values of ciprofloxacin (an organic antimicrobial compound inhibiting bacterial DNA topoisomerase) of 0.01, 0.15 and 0.12, respectively. On the other hand, the highest efficacy among the tested derivatives against C. albicans was recorded for the -NO2 substituent in position 2 (MIC = 0.31 μg/mL). The value was compared with clotrimazole (an organic compound with antifungal activity that inhibits the biosynthesis of sterols that build fungal cell membranes), for which the MIC value is 0.10 μg/mL [135].
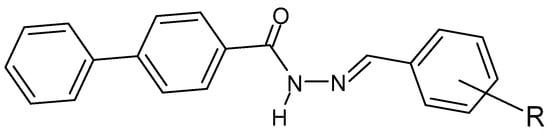
Figure 9. Hydrazide–hydrazone derivative of biphenyl-4-carboxylic acid, where R = NO2, -Cl or -Br [135].
The following compounds (Figure 10) were tested against streptomycin and against B. subtilis, K. pneumoniae and E. coli and showed lower MIC values for the chloro, fluoro and para substituents. Moreover, lower values were also recorded for the above-mentioned derivatives and for the derivative with the chlorine atom in the “ortho” position against fluconazole and against A. flavus, A. niger, C. albicans and Candida6 [136].
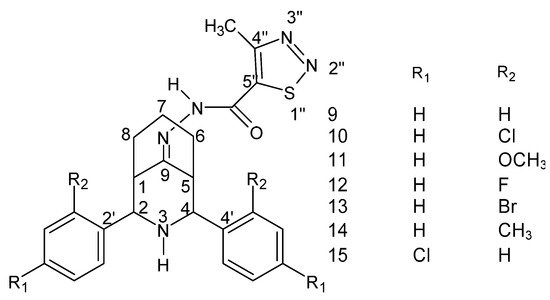
Figure 10. Structure of the 2r, 4c-diaryl-3-azabicyclo [3.3.1] nonan-9-one-4-methyl-1,2,3-thiadiazole-5-carbonyl hydrazide–hydrazone [136].
There have also been reports in the literature on the anti-cancer properties of hydrazide–hydrazones, e.g., in the case of colorectal cancer acting by increasing the permeability of the outer mitochondrial membranes of neoplastic cells. The cytotoxic activity of the hydrazide–hydrazone derivative of 2,6-difluorobenzoic acid and 6-bromoindole (Figure 11) was confirmed against the HTC-116, DLD-1 and SW-620 cell lines, while its lack—against healthy fibroblasts of the L929 cell line [77].
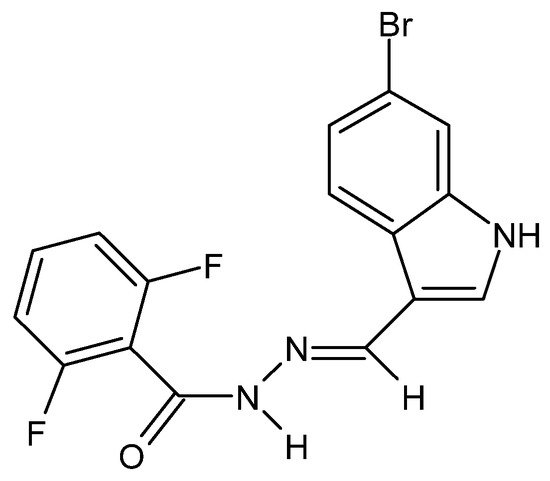
Figure 11. Derivative of 2,6-difluorobenzoic acid and 6-bromoindole—hydrazide–hydrazone with anti-cancer properties [77].
For the HTC-116 cell line, the pathway of programmed cell death has been established, starting with the inhibition of Bcl2 proteins, which regulate this process and act in an anti-apoptotic manner, and, consequently, regulate the release of pro-apoptotic cytochrome c. Caspases-9 and -3, which regulate the production of reactive oxygen species, take over further control of the process [77]. Cytochrome c binds Apaf-1, which is the first factor activating the apoptotic protease-activating factor 1. The formation of this complex (apoptosome) leads to the activation of procaspase-9 to the initiator caspase-9, which in turn activates the executive caspases, caspase-3 and caspase-7, of which caspase-3 is the master caspase and caspase-7 is the helper caspase. Caspase-9 begins the secretion of reactive oxygen species that have a proteolytic effect on the cell subjected to apoptosis, and caspase-3 acts as an inhibitor, ending the process [137].
Hydrazide–hydrazones are also used as antidiabetic agents in type II diabetes [138,139]. They play the role of non-competitive antagonists of the glucagon receptor—thus, they inhibit the glucagon-induced processes: glycogenolysis and gluconeogenesis, which in turn leads to a reduction in blood sugar levels.
A very interesting biological activity of the hydrazide–hydrazone of lactic acid was described by Noshiranzadeh et al. [140]. They synthesized a number of this type of lactic acid derivatives, two of which (Figure 12) showed particular activity against the selected bacterial strains (Minimum Inhibitory Concentration MIC = 64–128 µg/mL), which turned out to be lower than the reference gentamicin.
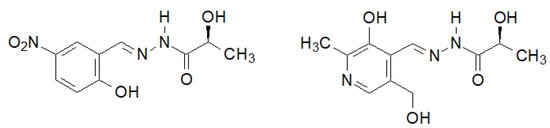
Figure 12. New hydrazide–hydrazones of lactic acid with antibacterial activity.
Olayinka et al. synthesized several 2-propylquinoline-4-carboxylic acid hydrazide–hydrazones [141]. The compound shown in the Figure 13 showed the highest activity against six strains of bacteria.
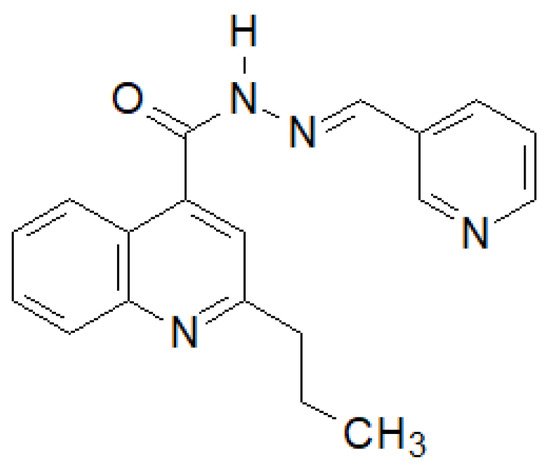
Figure 13. Quinoline derivative with significant antibacterial properties.
The authors showed that the presence of an electron-donating substituent in position 4 and an electron-withdrawing substituent in position 2 is of key importance for the activity of the derivatives obtained.
Indole-2-one was used by Salem et al. to obtain a series of hydrazide–hydrazones, of which the compound in Figure 14 had the most interesting properties [142].
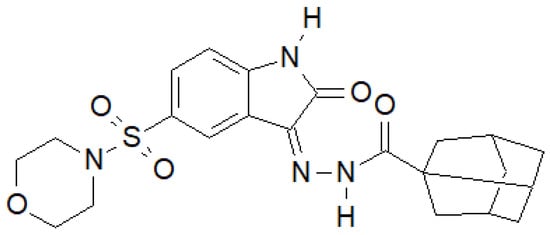
Figure 14. Indol-2-one derivative with antibacterial activity.
For the above compound, they performed an activity assay against DNA gyrase isolated from S. aureus, which showed that its half-maximal inhibitory concentration was IC50 = 19.32 ± 0.99 µM, while for the reference ciprofloxacin, IC50 = 26.43 ± 0.64 µM.
Of the N-substituted indole derivatives synthesized by Tiwari et al., the compound shown in Figure 15 showed the highest activity against Gram-positive bacteria [143]. In the case of E. coli MTCC 433 and B. subtilis MTCC 1427, it showed higher activity than the reference Chloramphenicol.
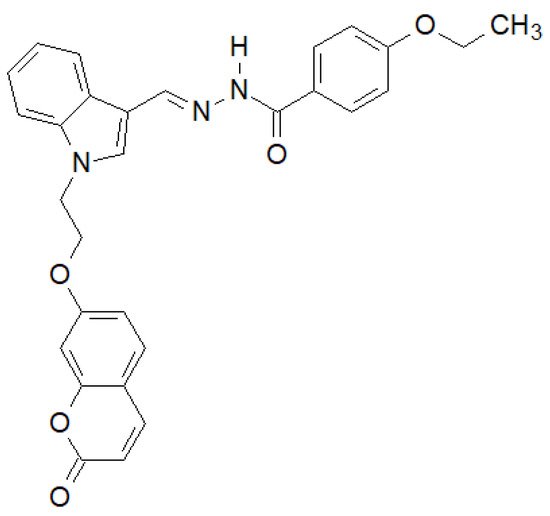
Figure 15. N-substituted indole derivative with antibacterial properties.
El-Etrawa et al. prepared a number of 2-thiouracil derivatives, of which the following compound (Figure 16) turned out to be the most active against E. coli, P. aeruginosa and S. aureus [144].
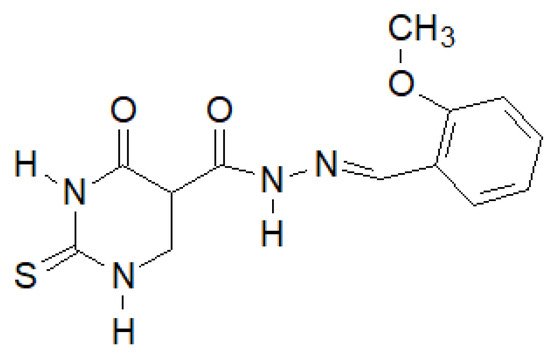
Figure 16. N-(2-Thiouracil-5-oyl)hydrazone derivative with antibacterial activity.
Recently, Paruch et al. synthesized 1,2,3-thiadiazole hydrazide–hydrazones, of which the compound shown in Figure 17 showed the highest activity [145].
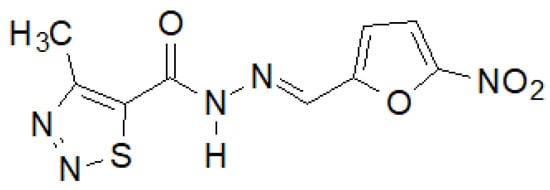
Figure 17. 4-Methyl-1,2,3-thiadiazole-carboxylic acid hydrazide derivative active against a panel of bacterial strains.
This compound showed activity against almost all tested bacterial strains, and in the case of S. epidermidis ATCC 12228 and M. luteus ATCC 10240, even two and eight times higher, respectively, than the reference nitrofurantoin.
In 2020, the results of the synthesis and biological tests of eugenol hydrazide–hydrazones were published [146]. The compound in the Figure 18 showed the highest activity against M. tuberculosis H37Rv.
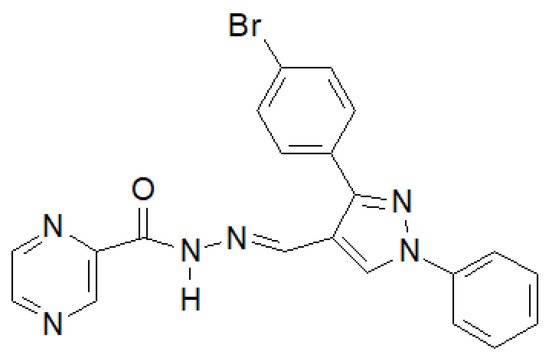
Figure 18. Pyrazine derivative with antitubercular properties.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27030787
This entry is offline, you can click here to edit this entry!